Valence Topological Charge-Transfer Indices for Dipole Moments: Percutaneous Enhancers

Abstract
:Introduction
Materials and methods
Valence charge-transfer indices for heteroatoms
Calculation Rresults and Discussion

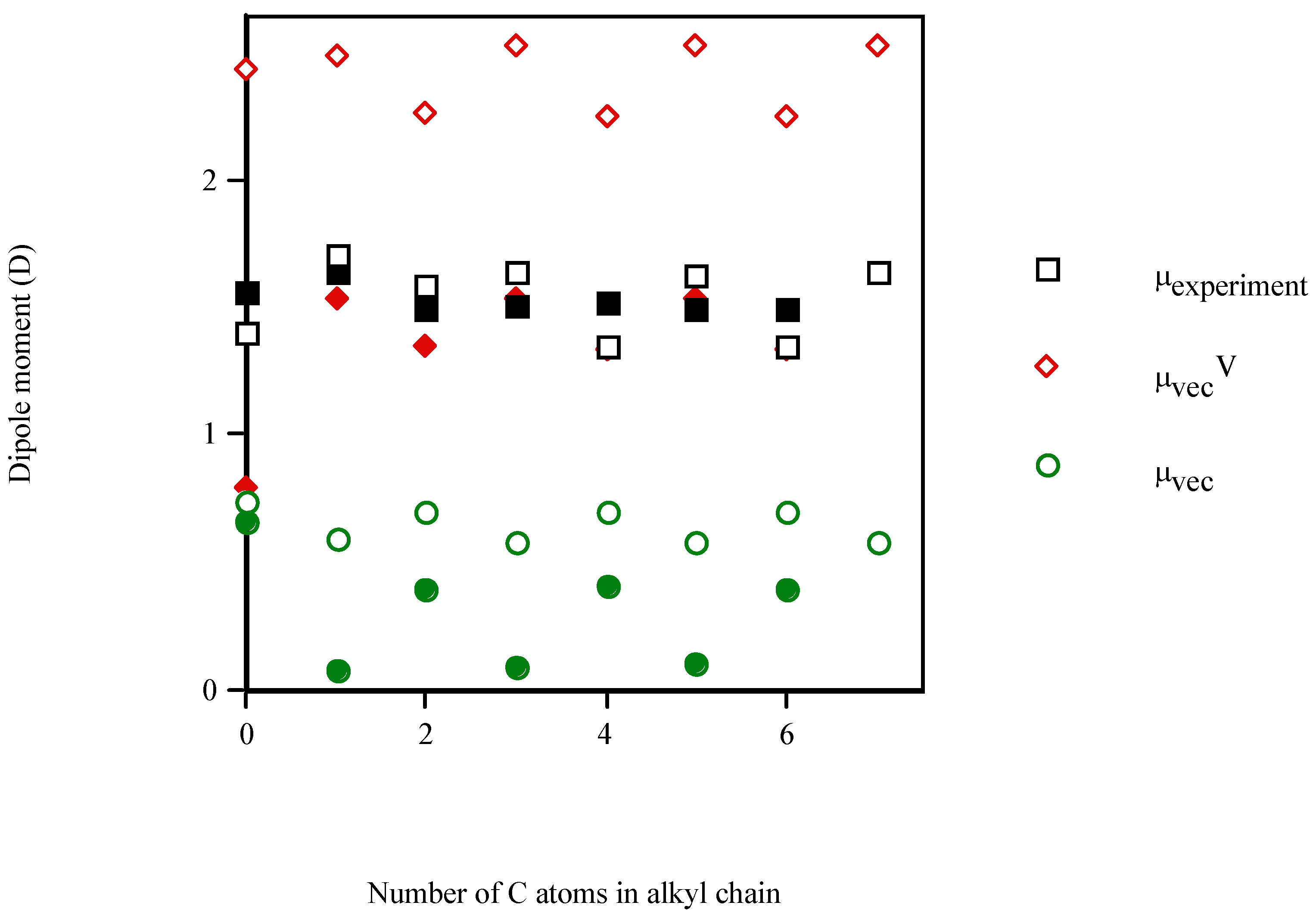{kind=link}
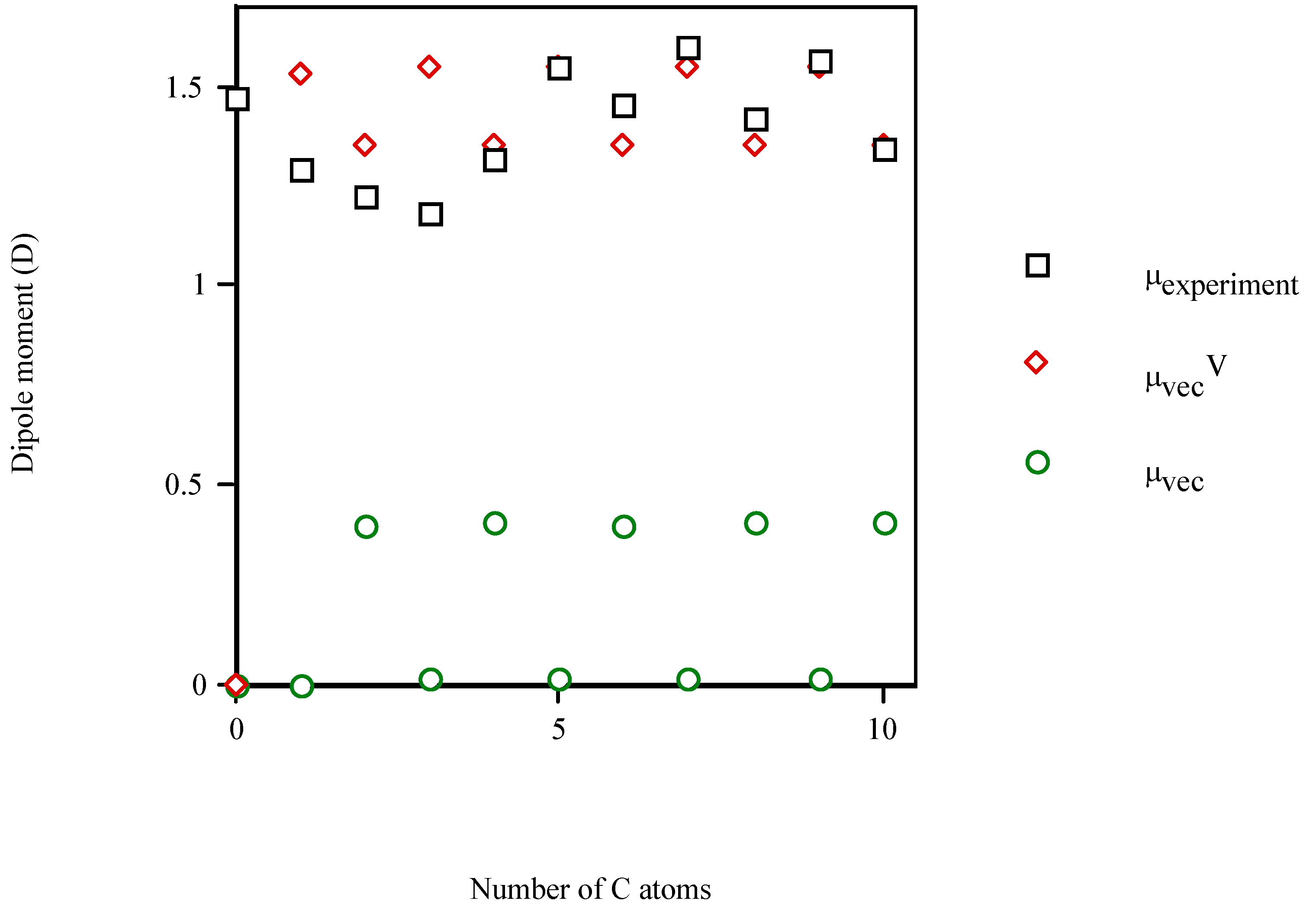{kind=link}
| Molecule | N | G1 | G2 | G3 | G4 | G5 |
|---|---|---|---|---|---|---|
| phenol | 7 | 2.0000 | 0.8889 | 0.3750 | 0.2222 | 0.0000 |
| benzyl alcohol | 8 | 1.2500 | 6.7778 | 0.8125 | 0.4133 | 0.1250 |
| 2‑phenyl‑1‑ethanol | 9 | 1.2500 | 6.7778 | 0.8750 | 0.5644 | 0.2431 |
| 3‑phenyl‑1‑propanol | 10 | 1.2500 | 6.7778 | 0.8750 | 0.6044 | 0.3333 |
| 4‑phenyl‑1‑butanol | 11 | 1.2500 | 6.7778 | 0.8750 | 0.6044 | 0.3611 |
| 5‑phenyl‑1‑pentanol | 12 | 1.2500 | 6.7778 | 0.8750 | 0.6044 | 0.3611 |
| 6‑phenyl‑1‑hexanol | 13 | 1.2500 | 6.7778 | 0.8750 | 0.6044 | 0.3611 |
| 7‑phenyl‑1‑heptanol | 14 | 1.2500 | 6.7778 | 0.8750 | 0.6044 | 0.3611 |
| 1‑phenyl‑2‑propanol | 10 | 2.2500 | 6.7778 | 0.9375 | 0.7156 | 0.3611 |
| 2‑phenyl‑2‑propanol | 10 | 2.7500 | 7.2222 | 1.5625 | 0.7956 | 0.3750 |
| 3‑phenyl‑2‑propen‑1‑ol | 10 | 1.2500 | 8.2222 | 0.8750 | 0.4844 | 0.2708 |
| 1‑phenyl‑1‑pentanol | 12 | 1.7500 | 7.1111 | 1.3125 | 0.8756 | 0.4861 |
| 1‑phenyl‑2‑pentanol | 12 | 2.2500 | 7.0000 | 1.0625 | 0.7556 | 0.4792 |
| aniline | 7 | 1.2500 | 6.6667 | 0.5000 | 0.2222 | 0.0000 |
| 4‑methylaniline | 8 | 2.5000 | 8.0000 | 0.8750 | 0.5244 | 0.0000 |
| 4‑ethylaniline | 9 | 2.5000 | 8.1111 | 1.1875 | 0.7156 | 0.1806 |
| 4‑propylaniline | 10 | 2.5000 | 8.1111 | 1.2500 | 0.8667 | 0.2986 |
| 4‑butylaniline | 11 | 2.5000 | 8.1111 | 1.2500 | 0.9067 | 0.3889 |
| 4‑pentylaniline | 12 | 2.5000 | 8.1111 | 1.2500 | 0.9067 | 0.4167 |
| 4‑hexylaniline | 13 | 2.5000 | 8.1111 | 1.2500 | 0.9067 | 0.4167 |
| 2‑methylaniline | 8 | 2.0000 | 8.2222 | 1.0000 | 0.4444 | 0.0000 |
| 4‑isopropylaniline | 10 | 3.0000 | 8.2222 | 1.5000 | 0.9067 | 0.3611 |
| 4‑(1’‑propenyl)aniline | 10 | 2.5000 | 9.4444 | 1.0000 | 0.7467 | 0.2639 |
| 1‑aminonaphthalene | 11 | 1.7500 | 12.2222 | 1.6875 | 0.9822 | 0.1250 |
| 2‑aminonaphthalene | 11 | 2.2500 | 12.0000 | 1.5000 | 0.9911 | 0.1736 |
| 4‑cyclohexylaniline | 13 | 2.5000 | 8.2222 | 1.5625 | 1.0578 | 0.5208 |
| 4‑aminobiphenyl | 13 | 2.2500 | 13.7778 | 1.2500 | 0.7911 | 0.4167 |
| Molecule | J1 | J2 | J3 | J4 | J5 | |
| phenol | 0.3333 | 0.1481 | 0.0625 | 0.0370 | 0.0000 | |
| benzyl alcohol | 0.1786 | 0.9683 | 0.1161 | 0.0590 | 0.0179 | |
| 2‑phenyl‑1‑ethanol | 0.1563 | 0.8472 | 0.1094 | 0.0706 | 0.0304 | |
| 3‑phenyl‑1‑propanol | 0.1389 | 0.7531 | 0.0972 | 0.0672 | 0.0370 | |
| 4‑phenyl‑1‑butanol | 0.1250 | 0.6778 | 0.0875 | 0.0604 | 0.0361 | |
| 5‑phenyl‑1‑pentanol | 0.1136 | 0.6162 | 0.0795 | 0.0549 | 0.0328 | |
| 6‑phenyl‑1‑hexanol | 0.1042 | 0.5648 | 0.0729 | 0.0504 | 0.0301 | |
| 7‑phenyl‑1‑heptanol | 0.0962 | 0.5214 | 0.0673 | 0.0465 | 0.0278 | |
| 1‑phenyl‑2‑propanol | 0.2500 | 0.7531 | 0.1042 | 0.0795 | 0.0401 | |
| 2‑phenyl‑2‑propanol | 0.3056 | 0.8025 | 0.1736 | 0.0884 | 0.0417 | |
| 3‑phenyl‑2‑propen‑1‑ol | 0.1389 | 0.9136 | 0.0972 | 0.0538 | 0.0301 | |
| 1‑phenyl‑1‑pentanol | 0.1591 | 0.6465 | 0.1193 | 0.0796 | 0.0442 | |
| 1‑phenyl‑2‑pentanol | 0.2045 | 0.6364 | 0.0966 | 0.0687 | 0.0436 | |
| aniline | 0.2083 | 1.1111 | 0.0833 | 0.0370 | 0.0000 | |
| 4‑methylaniline | 0.3571 | 1.1429 | 0.1250 | 0.0749 | 0.0000 | |
| 4‑ethylaniline | 0.3125 | 1.0139 | 0.1484 | 0.0894 | 0.0226 | |
| 4‑propylaniline | 0.2778 | 0.9012 | 0.1389 | 0.0963 | 0.0332 | |
| 4‑butylaniline | 0.2500 | 0.8111 | 0.1250 | 0.0907 | 0.0389 | |
| 4‑pentylaniline | 0.2273 | 0.7374 | 0.1136 | 0.0824 | 0.0379 | |
| 4‑hexylaniline | 0.2083 | 0.6759 | 0.1042 | 0.0756 | 0.0347 | |
| 2‑methylaniline | 0.2857 | 1.1746 | 0.1429 | 0.0635 | 0.0000 | |
| 4‑isopropylaniline | 0.3333 | 0.9136 | 0.1667 | 0.1007 | 0.0401 | |
| 4‑(1’‑propenyl)aniline | 0.2778 | 1.0494 | 0.1111 | 0.0830 | 0.0293 | |
| 1‑aminonaphthalene | 0.1750 | 1.2222 | 0.1688 | 0.0982 | 0.0125 | |
| 2‑aminonaphthalene | 0.2250 | 1.2000 | 0.1500 | 0.0991 | 0.0174 | |
| 4‑cyclohexylaniline | 0.2083 | 0.6852 | 0.1302 | 0.0881 | 0.0434 | |
| 4‑aminobiphenyl | 0.1875 | 1.1481 | 0.1042 | 0.0659 | 0.0347 | |
| Molecule | G1V | G2V | G3V | G4V | G5V | |
| phenol | 2.2000 | 1.1000 | 0.3639 | 0.0847 | 0.0000 | |
| benzyl alcohol | 2.9500 | 6.6611 | 0.5625 | 0.2533 | 0.0370 | |
| 2‑phenyl‑1‑ethanol | 2.9500 | 7.1056 | 0.7444 | 0.4044 | 0.1319 | |
| 3‑phenyl‑1‑propanol | 2.9500 | 7.1056 | 0.9944 | 0.5019 | 0.2222 | |
| 4‑phenyl‑1‑butanol | 2.9500 | 7.1056 | 0.9944 | 0.6619 | 0.2824 | |
| 5‑phenyl‑1‑pentanol | 2.9500 | 7.1056 | 0.9944 | 0.6619 | 0.3936 | |
| 6‑phenyl‑1‑hexanol | 2.9500 | 7.1056 | 0.9944 | 0.6619 | 0.3936 | |
| 7‑phenyl‑1‑heptanol | 2.9500 | 7.1056 | 0.9944 | 0.6619 | 0.3936 | |
| 1‑phenyl‑2‑propanol | 3.4500 | 7.6556 | 0.8069 | 0.5556 | 0.2500 | |
| 2‑phenyl‑2‑propanol | 3.4500 | 8.2056 | 1.3125 | 0.6356 | 0.2870 | |
| 3‑phenyl‑2‑propen‑1‑ol | 2.9500 | 8.3278 | 0.6306 | 0.3819 | 0.1597 | |
| 1‑phenyl‑1‑pentanol | 2.9500 | 7.3222 | 1.1819 | 0.7731 | 0.4861 | |
| 1‑phenyl‑2‑pentanol | 3.4500 | 7.6556 | 1.0514 | 0.7331 | 0.3681 | |
| aniline | 0.8500 | 6.2222 | 0.2556 | 0.1535 | 0.0000 | |
| 4‑methylaniline | 2.1000 | 7.5556 | 0.6306 | 0.4557 | 0.0440 | |
| 4‑ethylaniline | 2.1000 | 7.6667 | 0.9431 | 0.6468 | 0.1690 | |
| 4‑propylaniline | 2.1000 | 7.6667 | 1.0056 | 0.7979 | 0.2871 | |
| 4‑butylaniline | 2.1000 | 7.6667 | 1.0056 | 0.8379 | 0.3773 | |
| 4‑pentylaniline | 2.1000 | 7.6667 | 1.0056 | 0.8379 | 0.4051 | |
| 4‑hexylaniline | 2.1000 | 7.6667 | 1.0056 | 0.8379 | 0.4051 | |
| 2‑methylaniline | 1.6000 | 7.7778 | 0.8778 | 0.3757 | 0.0000 | |
| 4‑isopropylaniline | 2.6000 | 7.7778 | 1.2556 | 0.8379 | 0.3171 | |
| 4‑(1’‑propenyl)aniline | 2.1000 | 9.0000 | 0.7556 | 0.6779 | 0.2199 | |
| 1‑aminonaphthalene | 1.3500 | 11.6722 | 1.3208 | 0.7760 | 0.0810 | |
| 2‑aminonaphthalene | 1.8500 | 11.5556 | 1.2556 | 0.8536 | 0.0856 | |
| 4‑cyclohexylaniline | 2.1000 | 7.7778 | 1.3181 | 0.9890 | 0.4768 | |
| 4‑aminobiphenyl | 1.8500 | 13.3333 | 1.0056 | 0.7224 | 0.3727 | |
| Molecule | J1V | J2V | J3V | J4V | J5V | |
| phenol | 0.3637 | 0.1833 | 0.0606 | 0.0141 | 0.0000 | |
| benzyl alcohol | 0.4214 | 0.9516 | 0.0804 | 0.0362 | 0.0053 | |
| 2‑phenyl‑1‑ethanol | 0.3688 | 0.8882 | 0.0931 | 0.0506 | 0.0165 | |
| 3‑phenyl‑1‑propanol | 0.3278 | 0.7895 | 0.1105 | 0.0558 | 0.0247 | |
| 4‑phenyl‑1‑butanol | 0.2950 | 0.7106 | 0.0994 | 0.0662 | 0.0282 | |
| 5‑phenyl‑1‑pentanol | 0.2682 | 0.6460 | 0.0904 | 0.0602 | 0.0358 | |
| 6‑phenyl‑1‑hexanol | 0.2458 | 0.5921 | 0.0829 | 0.0552 | 0.0328 | |
| 7‑phenyl‑1‑heptanol | 0.2269 | 0.5466 | 0.0765 | 0.0509 | 0.0303 | |
| 1‑phenyl‑2‑propanol | 0.3833 | 0.8506 | 0.0897 | 0.0617 | 0.0278 | |
| 2‑phenyl‑2‑propanol | 0.3833 | 0.9117 | 0.1458 | 0.0706 | 0.0319 | |
| 3‑phenyl‑2‑propen‑1‑ol | 0.3278 | 0.9253 | 0.0701 | 0.0424 | 0.0177 | |
| 1‑phenyl‑1‑pentanol | 0.2682 | 0.6657 | 0.1074 | 0.0703 | 0.0442 | |
| 1‑phenyl‑2‑pentanol | 0.3136 | 0.6960 | 0.0956 | 0.0666 | 0.0335 | |
| aniline | 0.1417 | 1.0370 | 0.0426 | 0.0256 | 0.0000 | |
| 4‑methylaniline | 0.3000 | 1.0794 | 0.0901 | 0.0651 | 0.0063 | |
| 4‑ethylaniline | 0.2625 | 0.9583 | 0.1179 | 0.0809 | 0.0211 | |
| 4‑propylaniline | 0.2333 | 0.8519 | 0.1117 | 0.0887 | 0.0319 | |
| 4‑butylaniline | 0.2100 | 0.7667 | 0.1006 | 0.0838 | 0.0377 | |
| 4‑pentylaniline | 0.1909 | 0.6970 | 0.0914 | 0.0762 | 0.0368 | |
| 4‑hexylaniline | 0.1750 | 0.6389 | 0.0838 | 0.0698 | 0.0338 | |
| 2‑methylaniline | 0.2286 | 1.1111 | 0.1254 | 0.0537 | 0.0000 | |
| 4‑isopropylaniline | 0.2889 | 0.8642 | 0.1395 | 0.0931 | 0.0352 | |
| 4‑(1’‑propenyl)aniline | 0.2333 | 1.0000 | 0.0840 | 0.0753 | 0.0244 | |
| 1‑aminonaphthalene | 0.1350 | 1.1672 | 0.1321 | 0.0776 | 0.0081 | |
| 2‑aminonaphthalene | 0.1850 | 1.1556 | 0.1256 | 0.0854 | 0.0086 | |
| 4‑cyclohexylaniline | 0.1750 | 0.6481 | 0.1098 | 0.0824 | 0.0397 | |
| 4‑aminobiphenyl | 0.1542 | 1.1111 | 0.0838 | 0.0602 | 0.0311 |
| Molecule | Number of carbon atoms in alkyl chain | Vector semisum | Valence vector semisum | Experimentala |
|---|---|---|---|---|
| phenol | 0 | 0.737 | 2.431 | 1.400 (1.233b) |
| benzyl alcohol | 1 | 0.589 | 2.487 | 1.700 (1.568b) |
| 2‑phenyl‑1‑ethanol | 2 | 0.700 | 2.257 | 1.590 (1.497b) |
| 3‑phenyl‑1‑propanol | 3 | 0.573 | 2.519 | 1.640 (1.597b) |
| 4‑phenyl‑1‑butanol | 4 | 0.702 | 2.249 | 1.345b |
| 5‑phenyl‑1‑pentanol | 5 | 0.573 | 2.519 | 1.626b |
| 6‑phenyl‑1‑hexanol | 6 | 0.702 | 2.250 | 1.346b |
| 7‑phenyl‑1‑heptanol | 7 | 0.573 | 2.518 | 1.634b |
| 1‑phenyl‑2‑propanol | 3 | 0.923 | 2.347 | 1.564b |
| 2‑phenyl‑2‑propanol | 3 | 0.780 | 2.821 | 1.463b |
| 3‑phenyl‑2‑propen‑1‑ol | 3 | 0.561 | 2.495 | 1.591b |
| 1‑phenyl‑1‑pentanol | 5 | 0.426 | 2.794 | 1.746b |
| 1‑phenyl‑2‑pentanol | 5 | 0.895 | 2.367 | 1.496b |
| aniline | 0 | 0.655 | 0.792 | 1.560 (1.584b) |
| 4‑methylaniline | 1 | 0.079 | 1.526 | 1.640 (1.423b) |
| 4‑ethylaniline | 2 | 0.385 | 1.339 | 1.492b |
| 4‑propylaniline | 3 | 0.087 | 1.533 | 1.502b |
| 4‑butylaniline | 4 | 0.398 | 1.333 | 1.511b |
| 4‑pentylaniline | 5 | 0.093 | 1.533 | 1.489b |
| 4‑hexylaniline | 6 | 0.395 | 1.332 | 1.496b |
| 2‑methylaniline | 1 | 1.187 | 0.779 | 1.590 (1.560b) |
| 4‑isopropylaniline | 3 | 0.625 | 1.490 | 1.450b |
| 4‑(1’‑propenyl)aniline | 3 | 0.108 | 1.512 | 1.512b |
| 1‑aminonaphthalene | 4 | 0.645 | 0.802 | 1.490 (1.690b) |
| 2‑aminonaphthalene | 4 | 0.655 | 0.792 | 1.820 (1.868b) |
| 4‑cyclohexylaniline | 6 | 0.480 | 1.126 | 1.760 (1.447b) |
| 4‑aminobiphenyl | 6 | 0.651 | 0.795 | 1.447b |
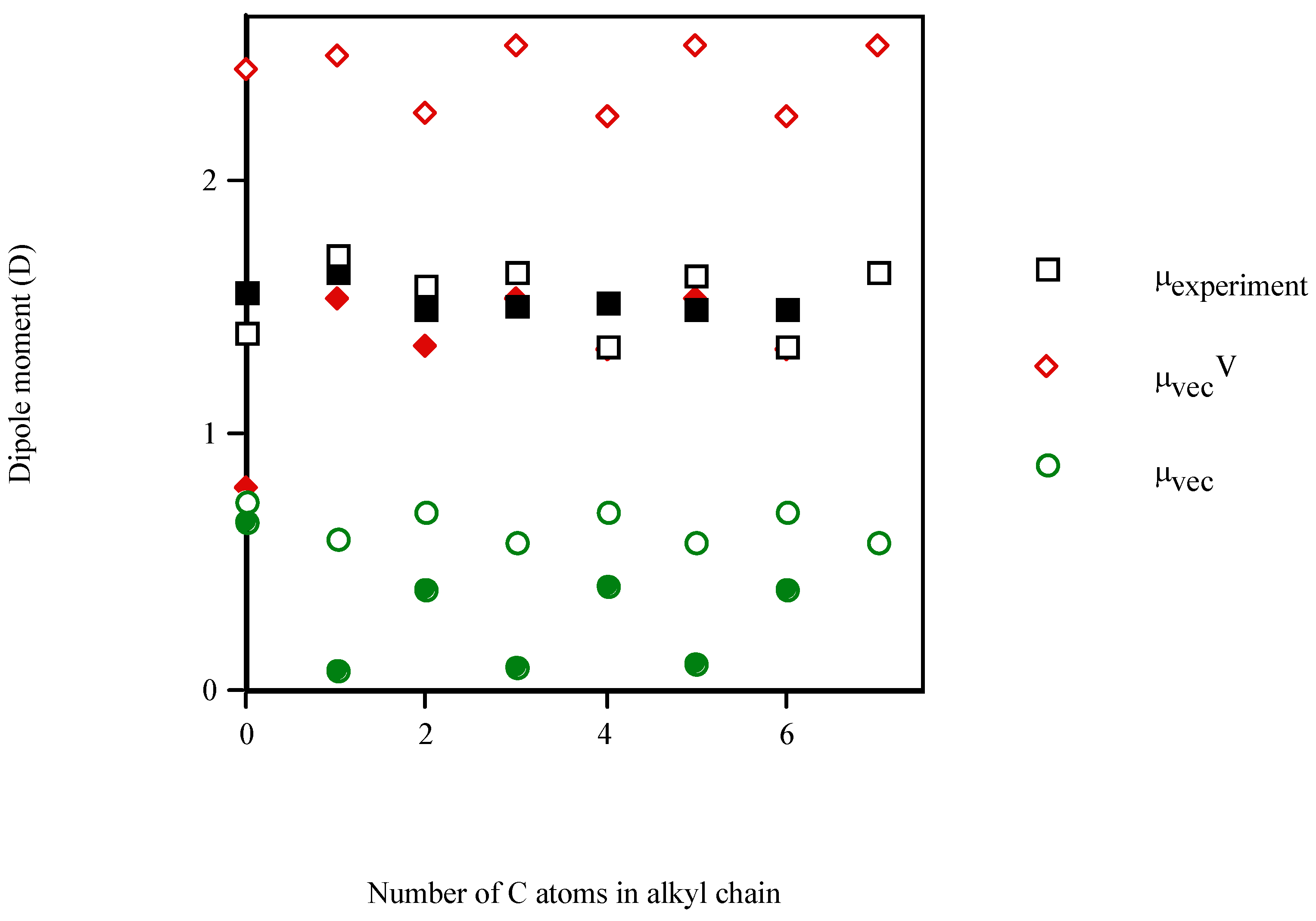
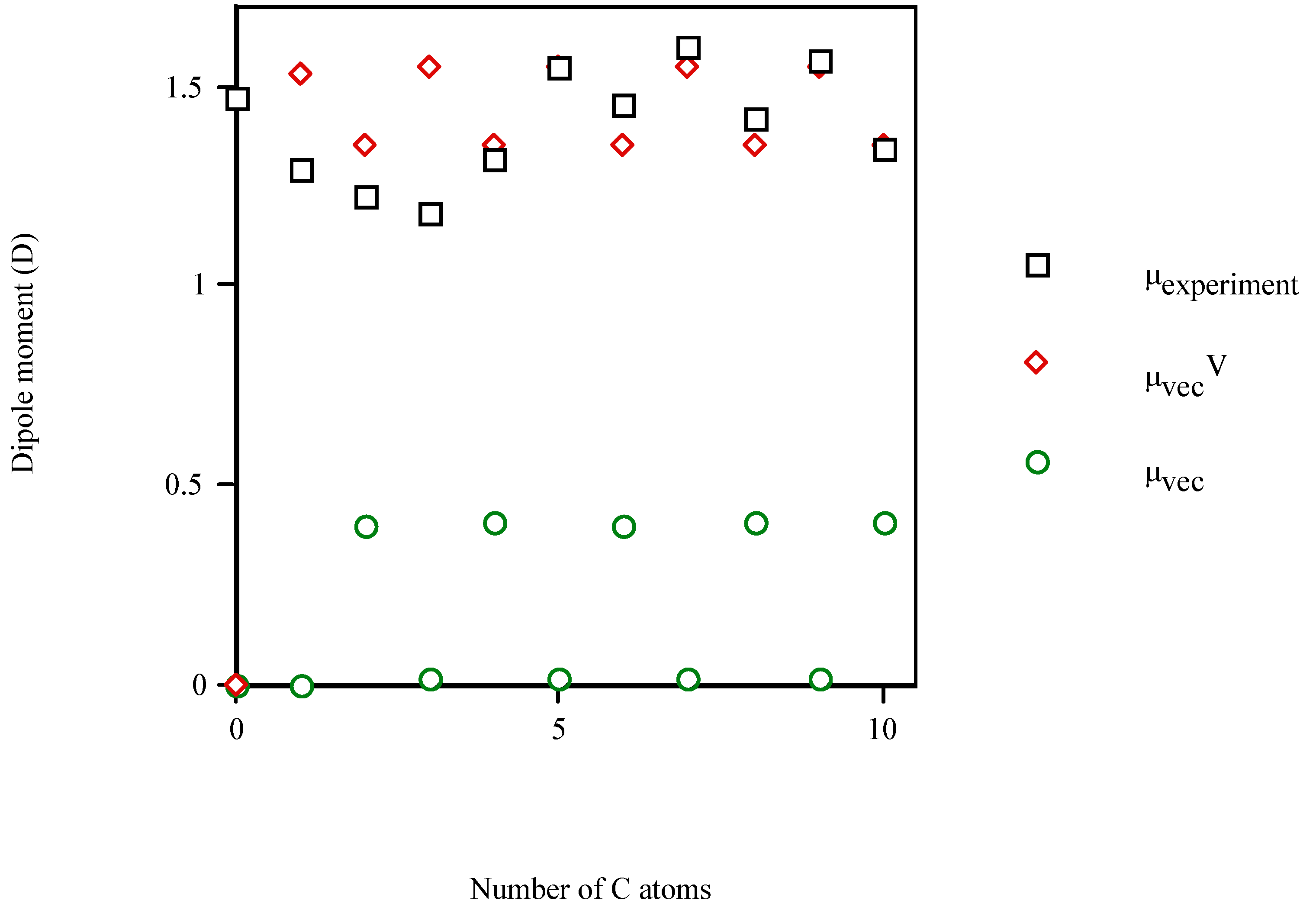
Conclusions
- Inclusion of the heteroatom in the π‑electron system was beneficial for the description of the dipole moment, owing to either the role of the additional p orbitals provided by the heteroatom or the role of steric factors in the π‑electron conjugation. The analysis of the electronic and steric factors in μ caused by the heteroatom showed that both factors are antagonistic, and that the electronic factor dominates over the steric one.
- The conjugated double bond in the alkyl chain of 3‑phenyl‑2‑propen‑1‑ol and 4‑(1’‑propenyl)-aniline lent to more rigid structures with dipole moment variations <1%.
- Linear and quadratic correlation models were obtained for the molecular dipole moments of phenyl alcohols, 4‑alkylanilines and aliphatic amines. μvecV improved the multivariable regression equations for μ, diminishing the risk of co‑linearity in the fit. Improvements in correlation suggest the general applicability of this index.
Acknowledgements
References
- Stehle, R.G.; Higuchi, W.I. In Vitro Model for Transport of Solutes in Three-Phase System II: Experimental Considerations. J. Pharm. Sci. 1972, 61, 1931–1935. [Google Scholar] [CrossRef]
- Lovering, E.G.; Black, D.B. Diffusion Layers Effects on Permeation of Phenylbutazone through Polydimethylsiloxane. J. Pharm. Sci. 1974, 63, 1399–1402. [Google Scholar] [CrossRef]
- Barry, B.W. Novel Mechanisms and Devices to Enable Successful Transdermal Drug Delivery. Eur. J. Pharm. Sci. 2001, 14, 101–114. [Google Scholar] [CrossRef]
- Sato, S.; Kim, S.W. Macromolecular Diffusion through Polymer Membranes. Int. J. Pharm. 1984, 22, 229–255. [Google Scholar] [CrossRef]
- Ackermann, C.; Flynn, G.L.; van Wyk, C.J. Percutaneous Absoption of Urea. Int. J. Cosm. Sci. 1985, 7, 251–264. [Google Scholar] [CrossRef]
- Ackermann, C.; Flynn, G.L.; Smith, W.M. Ether-Water Partitioning and Permeability through Nude Mouse Skin in Vitro. II. Hydrocortisone‑n‑alkyl Esters, Alkanols and Hydrophilic Compounds. Int. J. Pharm. 1987, 36, 67–71. [Google Scholar] [CrossRef]
- Ackermann, C.; Flynn, G.L. Ether-Water Partitioning and Permeability through Nude Mouse Skin in Vitro. I. Urea, Thiourea, Glycerol and Glucose. Int. J. Pharm. 1987, 36, 61–66. [Google Scholar] [CrossRef]
- Díez-Sales, O.; Copoví, A.; Casabó, V.G.; Herráez, M. A Modelistic Approach Showing the Importance of the Stagnant Aqueous Layers in in Vitro Diffusion Studies, and in Vitro-in Vivo Correlations. Int. J. Pharm. 1991, 77, 1–11. [Google Scholar]
- Plá-Delfina, J.M.; Pérez-Buendía, M.D.; Casabó, V.G.; Peris-Ribera, J.E.; Sánchez-Moyano, E.; Martín-Villodre, A. Absorption Partition Relationships for True Homologous Series of Xenobiotics as a Possible Approach to Study Mechanisms of Surfactants in Absorption. I. Aromatic Amines in Rat Colon. Int. J. Pharm. 1987, 37, 49–64. [Google Scholar]
- Collado, E.F.; Fabra-Campos, S.; Peris-Ribera, J.E.; Casabó, V.G.; Martín-Villodre, A.; Plá-Delfina, J.M. Absorption Partition Relationships for True Homologous Series of Xenobiotics as a Possible Approach to Study Mechanisms of Surfactants in Absorption. II. Aromatic Amines in Rat Small Intestine. Int. J. Pharm. 1988, 44, 187–196. [Google Scholar]
- Garrigues, T.M.; Collado, E.F.; Fabra-Campos, S.; Pérez-Buendía, M.D.; Martín-Villodre, A.; Plá-Delfina, J.M. Absorption Partition Relationships for True Homologous Series of Xenobiotics as a Possible Approach to Study Mechanisms of Surfactants in Absorption. III. Aromatic Amines and Cationic Surfactants. Int. J. Pharm. 1989, 57, 189–196. [Google Scholar]
- Garrigues, T.M.; Pérez-Varona, A.T.; Climent, E.; Bermejo, M.V.; Martín-Villodre, A.; Plá-Delfina, J.M. Gastric Absorption of Acidic Xenobiotics in the Rat: Biophysical Interpretation of an Apparently Atypical Behaviour. Int. J. Pharm. 1990, 64, 127–138. [Google Scholar]
- Díez-Sales, O.; Guzmán, D.; Cano, D.; Martín, A.; Sánchez, E.; Herráez, M. A Comparative in Vitro Study of Permeability with Different Synthetic and Biological Membranes. Eur. J. Drug Metab. Pharmacokinet. 1991, Spec. No. 3. 441–446. [Google Scholar]
- Sánchez-Moyano, E.; Seco, C.; Santolaria, A.; Fabra-Campos, S.; Herráez, M.; Matín-Villodre, A. Partition Behavior of Anilines in Bulk-Phase and High-Performance Liquid Chromatographic Systems: Influence on Correlation with Biological Constants. J. Pharm. Sci. 1992, 81, 720–725. [Google Scholar]
- Díez-Sales, O.; López-Castellano, A.; Maiques-Lacer, F.J.; Herráez-Domínguez, M. An in Vitro Percutaneous Absorption Study of Non-ionic Compounds across Human Skin. Pharmazie 1993, 48, 684–686. [Google Scholar]
- Díez-Sales, O.; Pérez-Sayas, E.; Martín-Villodre, A.; Herráez-Domínguez, M. The Prediction of Percutaneous Absorption: I. Influence of the Dermis on in Vitro Permeation Models. Int. J. Pharm. 1993, 100, 1–7. [Google Scholar]
- Díez-Sales, O.; Watkinson, A.C.; Herráez-Domínguez, M.; Javaloyes, C.; Hadgraft, J. A Mechanistic Investigation of the in-Vitro Human Skin Permeation Enhancing Effect of Atone®. Int. J. Pharm. 1996, 129, 33–40. [Google Scholar]
- López, A.; Morant, M.J.; Guzmán, D.; Borrás-Blasco, J.; Díez-Sales, O.; Herráez, M. Skin Permeation Model of Phenylalkylcarboxylic Homologous Acids and their Enhancer Effect on Percutaneous Penetration of 5-Fluorouracil. Int. J. Pharm. 1996, 139, 205–213. [Google Scholar]
- López, A.; Pellett, M.A.; Llinares, F.; Díez-Sales, O.; Herráez, M.; Hadgraft, J. The Enhancer Effect of Several Phenyl Alcohols on Percutaneous Penetration of 5-Fluorouracil. Pharm. Res. 1997, 14, 681–685. [Google Scholar]
- López, A.; Faus, V.; Díez-Sales, O.; Herráez, M. Skin Permeation Model of Phenyl Alcohols: Comparison of Experimental Conditions. Int. J. Pharm. 1998, 173, 183–191. [Google Scholar]
- Yalkowsky, S.H.; Flynn, G.L. Transport of Alkyl Homologs across Synthetic and Biological Membranes: A New Model for Chain Length–Activity Relationships. J. Pharm. Sci. 1973, 62, 210–217. [Google Scholar] [CrossRef]
- Flynn, G.L.; Yalkowsky, S.H.; Roseman, T.J. Mass Transport Phenomena and Models: Theoretical Concepts. J. Pharm. Sci. 1974, 63, 479–510. [Google Scholar] [CrossRef]
- Irwin, W.J.; Sanderson, F. D.; Po, A.L.W. Percutaneous Absorption of Ibuprofen: Vehicle Effects on Transport through Rat Skin. Int. J. Pharm. 1990, 66, 193–200. [Google Scholar] [CrossRef]
- Swarbrick, J.; Lee, G.; Brom, J.; Gensmantel, N.P. Drug Permeation through Human Skin II: Permeability of Ionizable Compounds. J. Pharm. Sci. 1984, 73, 1352–1355. [Google Scholar] [CrossRef]
- Banerjee, P.S.; Ritschel, W.A. Transdermal Permeation of Vasopressin. I. Influence of pH, Concentration, Shaving and Surfactant on in Vitro Permeation. Int. J. Pharm. 1989, 49, 189–197. [Google Scholar] [CrossRef]
- Lewis, D.; Hadgraft, J. Mixed Monolayers of Dipalmitoylphosphatidylcholine with Azone or Oleic Acid at the Air-Water Interface. Int. J. Pharm. 1990, 65, 211–218. [Google Scholar] [CrossRef]
- Williams, A.C.; Barry, B.W. Terpenes and the Lipid–Protein Partitioning Theory of Skin Penetration Enhancement. Pharm. Res. 1991, 8, 17–24. [Google Scholar]
- Torrens, F. A New Topological Index to Elucidate Apolar Hydrocarbons. J. Comput.-Aided Mol. Des. 2001, 15, 709–719. [Google Scholar] [CrossRef]
- Torrens, F. Valence Topological Charge-Transfer Indices for Dipole Moments. J. Mol. Struct. (Theochem) 2003, 621, 37–42. [Google Scholar] [CrossRef]
- Torrens, F. Valence Topological Charge-Transfer Indices for Dipole Moments. Mol. Diversity. in press.
- Torrens, F. Valence Topological Charge-Transfer Indices for Reflecting Polarity: Correction for Heteromolecules. Molecules. submitted for publication.
- Torrens, F. Valence Topological Charge-Transfer Indices for Dipole Moments. Molecules 2003, 8, 169–185. [Google Scholar] [CrossRef]
- Torrens, F. A New Chemical Index Inspired by Biological Plastic Evolution. Indian J. Chem., Sect. A 2003, 42, 1258–1263. [Google Scholar]
- Torrens, F. A Chemical Index Inspired by Biological Plastic Evolution: Valence-Isoelectronic Series of Aromatics. J. Chem. Inf. Comput. Sci. 2004, 44, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Torrens, F. Fractal Dimension of Transdermal-Delivery Drug Models. Leb. Sci. J. in press.
- Torrens, F. Fractal Dimension of Transdermal-Delivery Drug Models: 4‑Alkylanilines. Physica A. submitted for publication.
- Randić, M. On Characterization of Molecular Branching. J. Am. Chem. Soc. 1975, 97, 6609–6615. [Google Scholar] [CrossRef]
- Hosoya, H. Topological Index. A Newly Proposed Quantity Characterizing the Topological Nature of Structural Isomers of Saturated Hydrocarbons. Bull. Chem. Soc. Jpn. 1971, 44, 2332–2339. [Google Scholar] [CrossRef]
- Gálvez, J.; García, R.; Salabert, M.T.; Soler, R. Charge Indexes. New Topological Descriptors. J. Chem. Inf. Comput. Sci. 1984, 34, 520–525. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. Molecular Connectivity VII: Specific Treatment of Heteroatoms. J. Pharm. Sci. 1976, 65, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Lakard, B. Analysis of Permanent Electric Dipole Moments of Aliphatic Amines. Internet Electron. Conference of Molecular Design 2003. [Google Scholar]
- Box, G.E.P.; Hunter, W.G.; MacGregor, J.F.; Erjavec, J. Some Problems Associated with the Analysis of Multiresponse Data. Technometrics 1973, 15, 33–51. [Google Scholar] [CrossRef]
- Hocking, R.R. The Analysis and Selection of Variables in Linear Regression. Biometrics 1976, 32, 1–49. [Google Scholar]
- McClellan, A.L. Tables of Experimental Dipole Moments; Freeman: San Francisco, 1963. [Google Scholar]
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Torrens, F. Valence Topological Charge-Transfer Indices for Dipole Moments: Percutaneous Enhancers. Molecules 2004, 9, 1222-1235. https://doi.org/10.3390/91201222
Torrens F. Valence Topological Charge-Transfer Indices for Dipole Moments: Percutaneous Enhancers. Molecules. 2004; 9(12):1222-1235. https://doi.org/10.3390/91201222
Chicago/Turabian StyleTorrens, Francisco. 2004. "Valence Topological Charge-Transfer Indices for Dipole Moments: Percutaneous Enhancers" Molecules 9, no. 12: 1222-1235. https://doi.org/10.3390/91201222