A Novel Strategy Towards the Asymmetric Synthesis of Orthogonally Funtionalised 2-N-Benzyl-N-?-methylbenzylamino- 5-carboxymethyl-cyclopentane-1-carboxylic acid.

{kind=link}
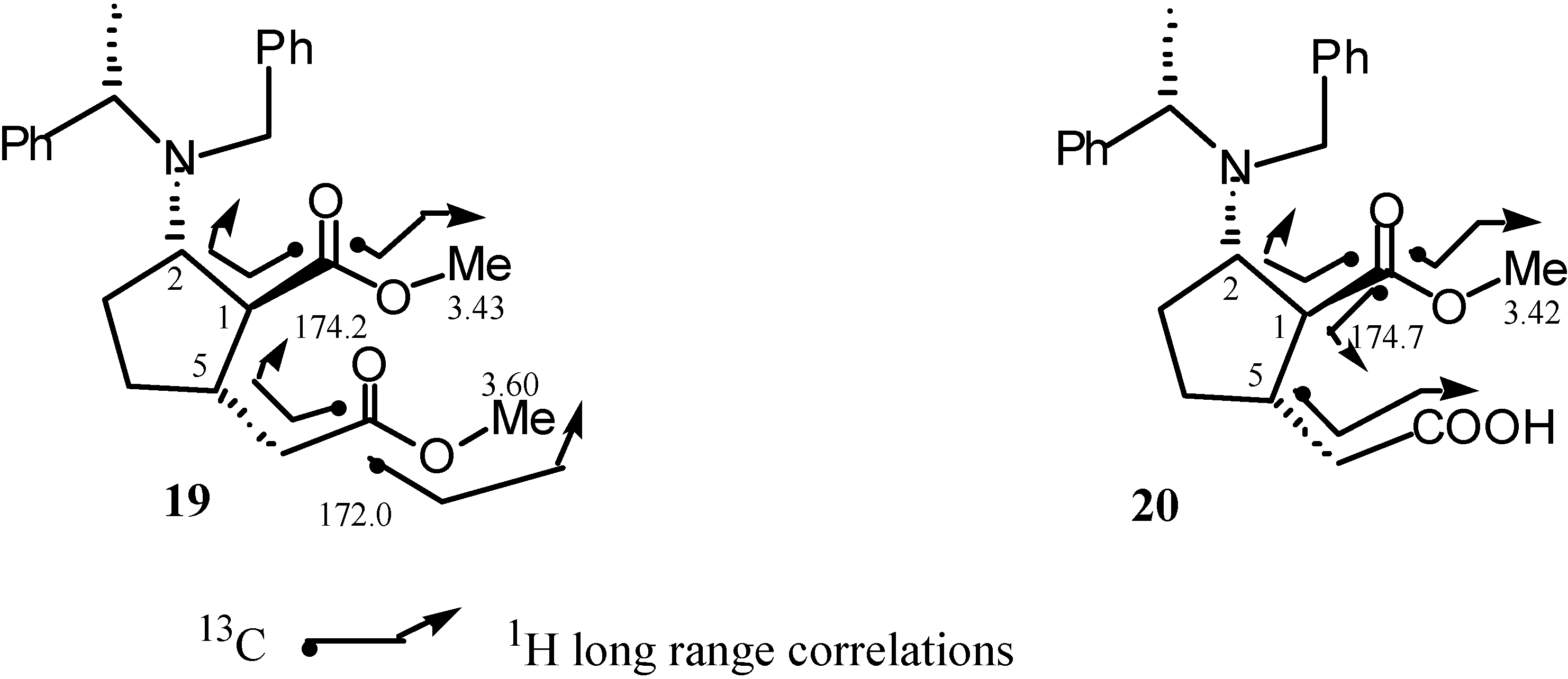{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
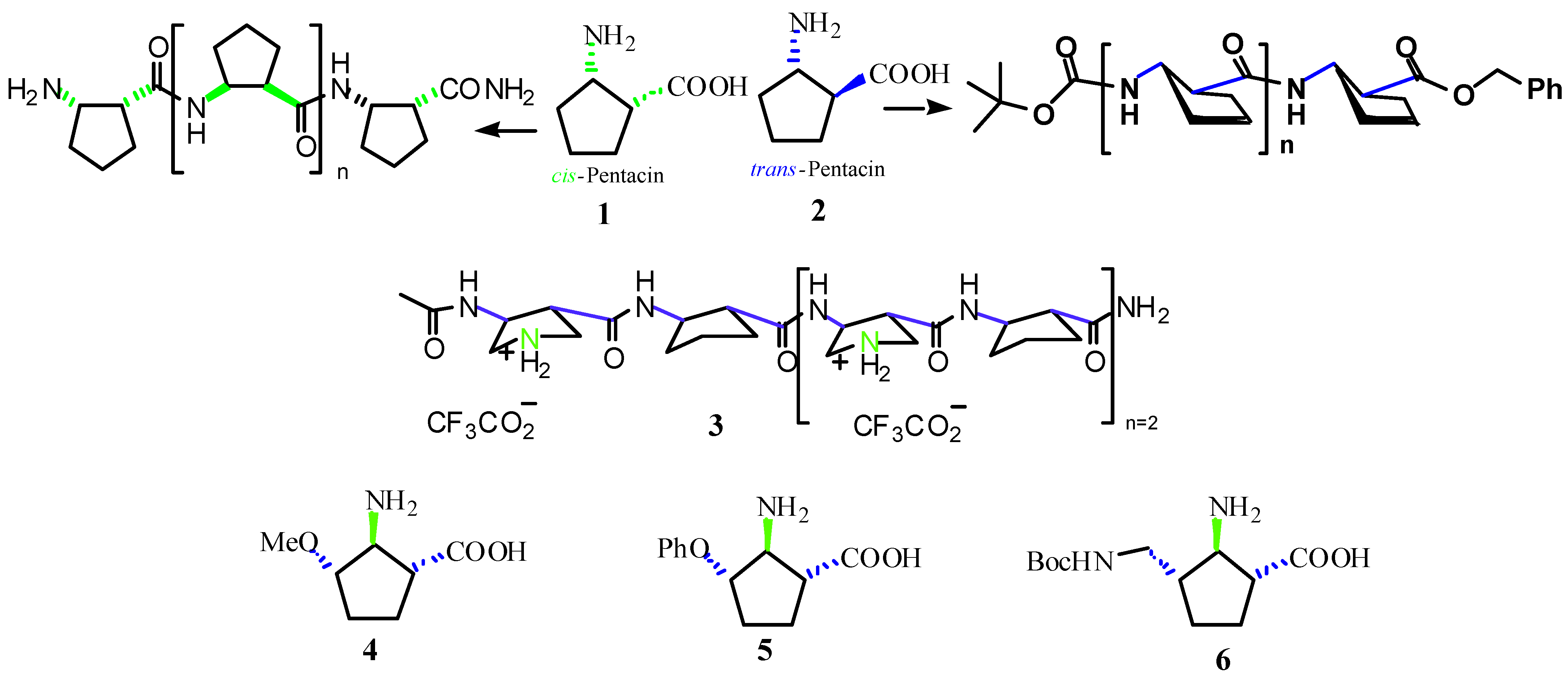
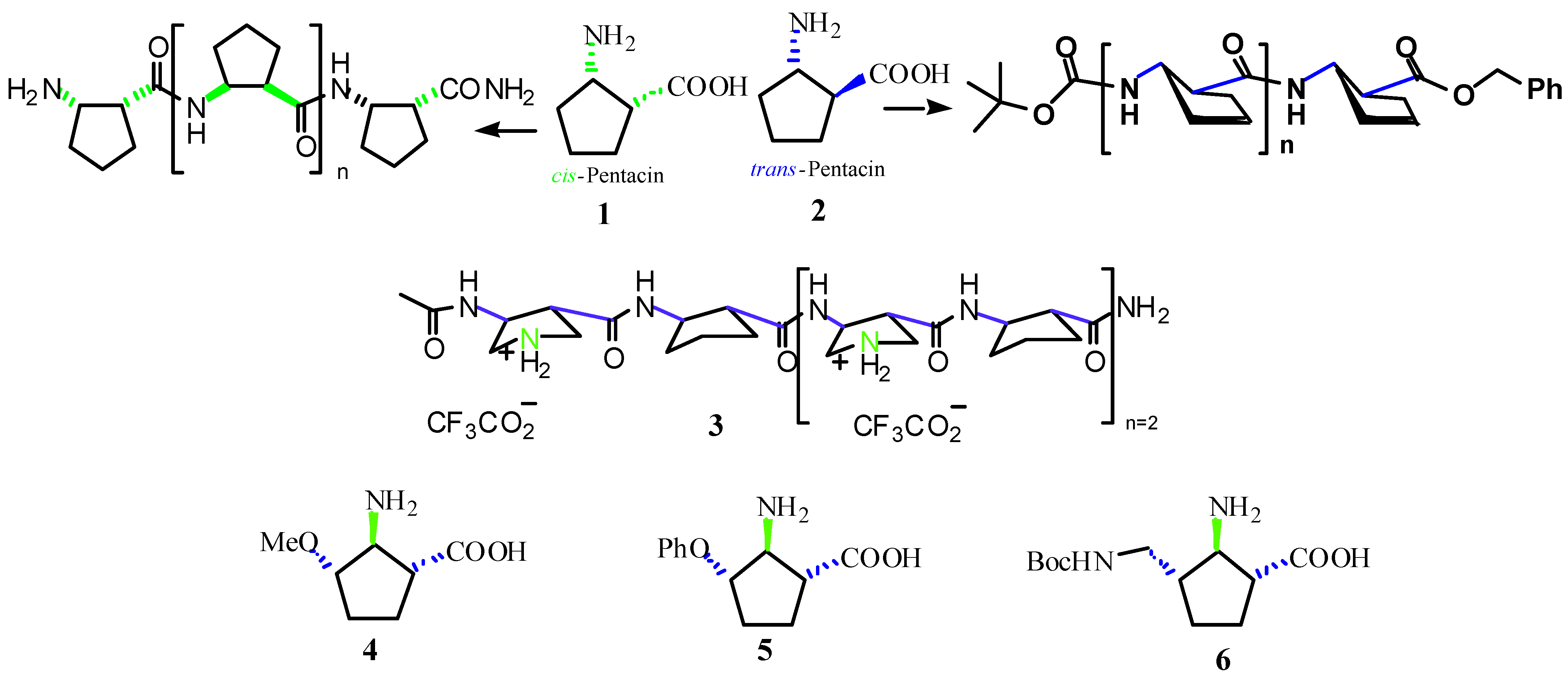
:Introduction
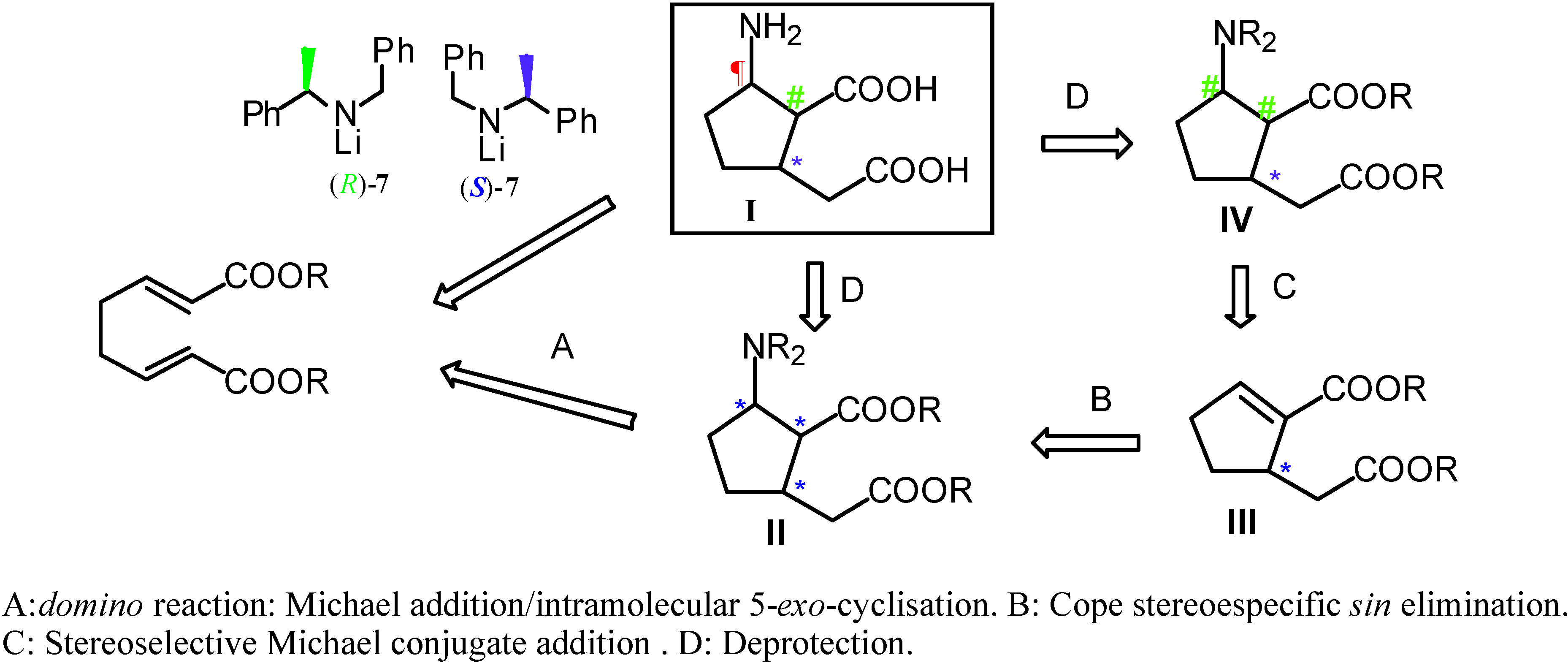
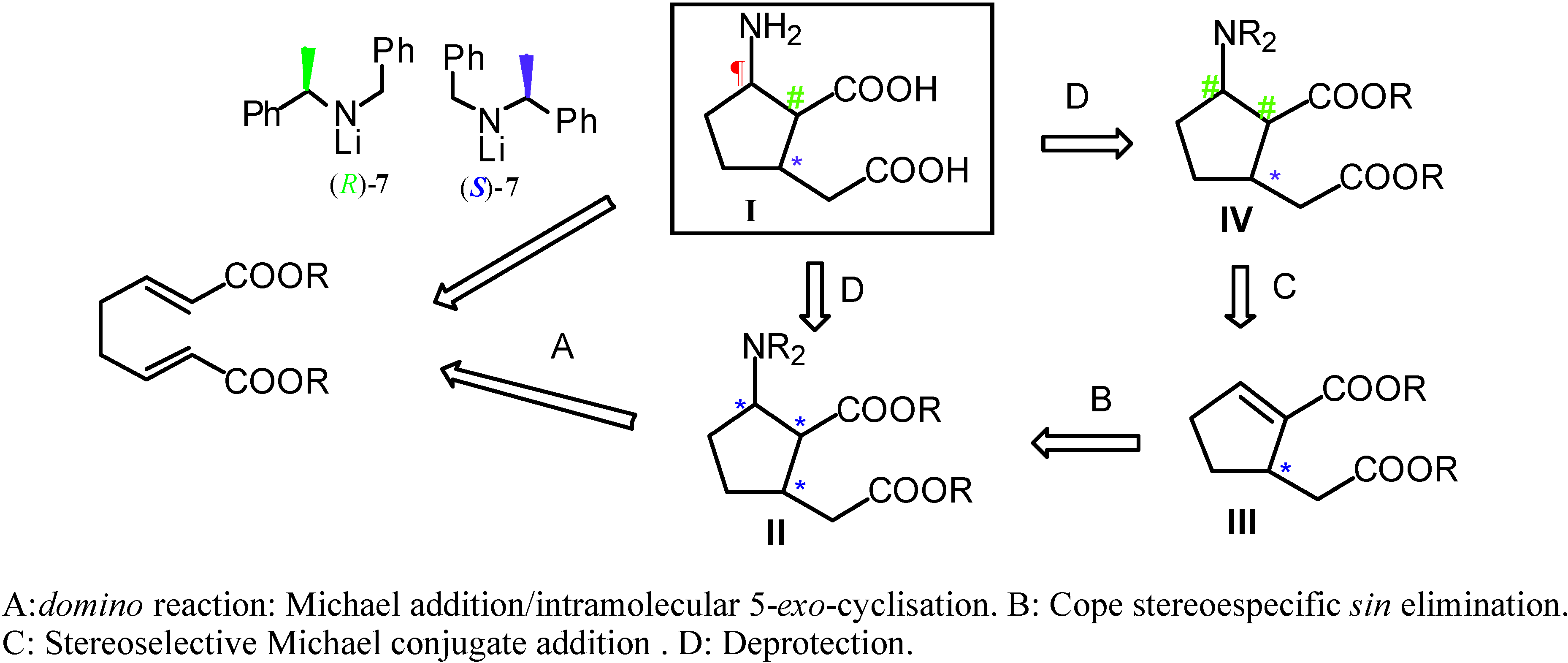
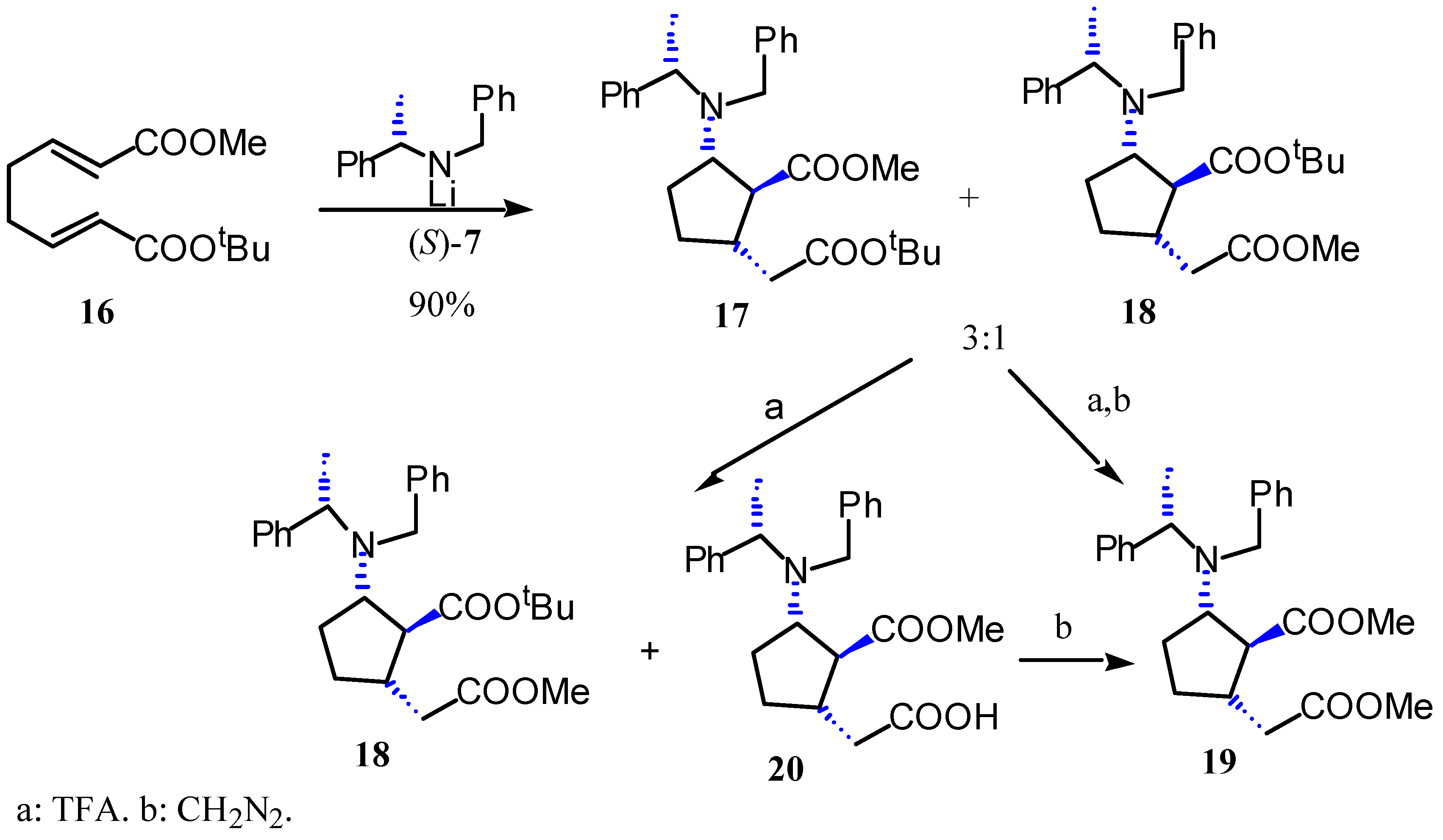
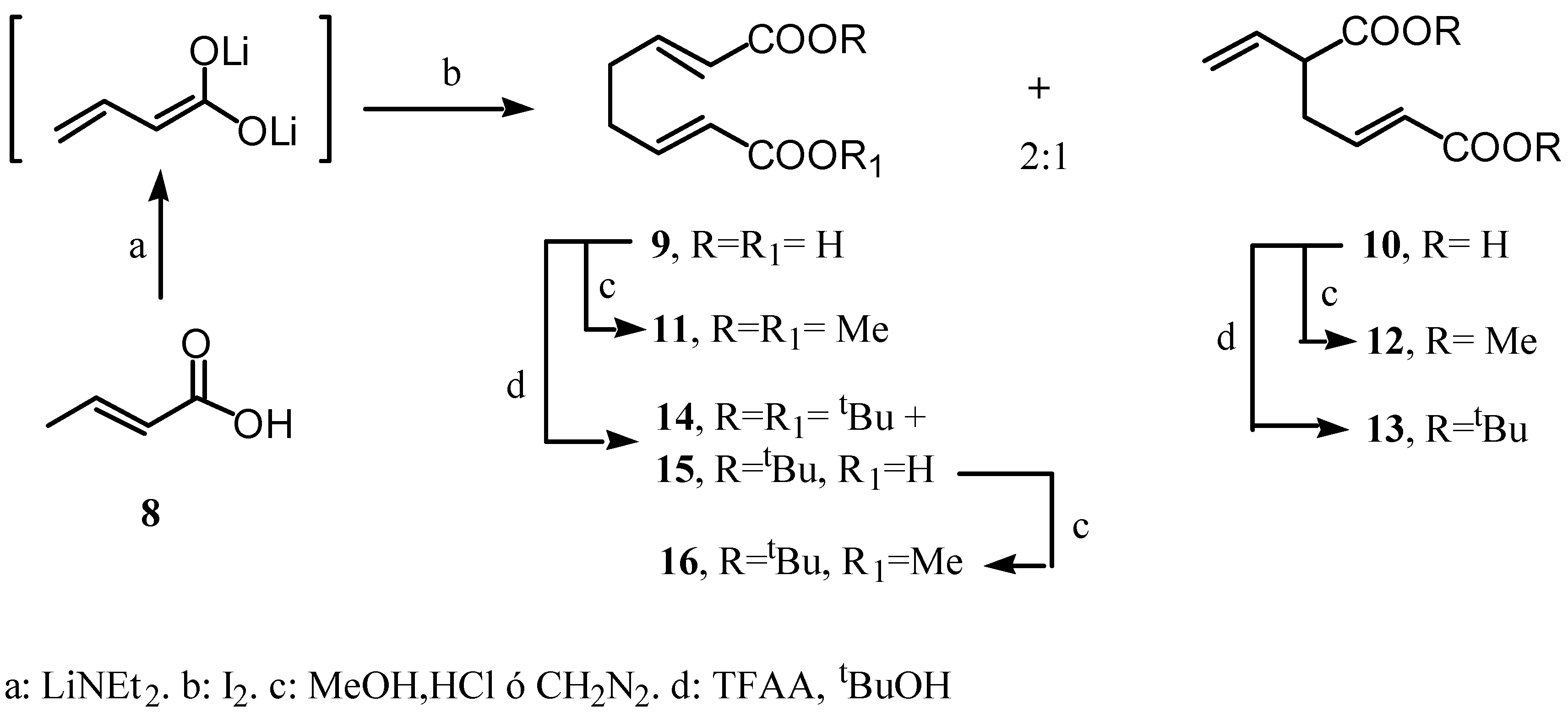
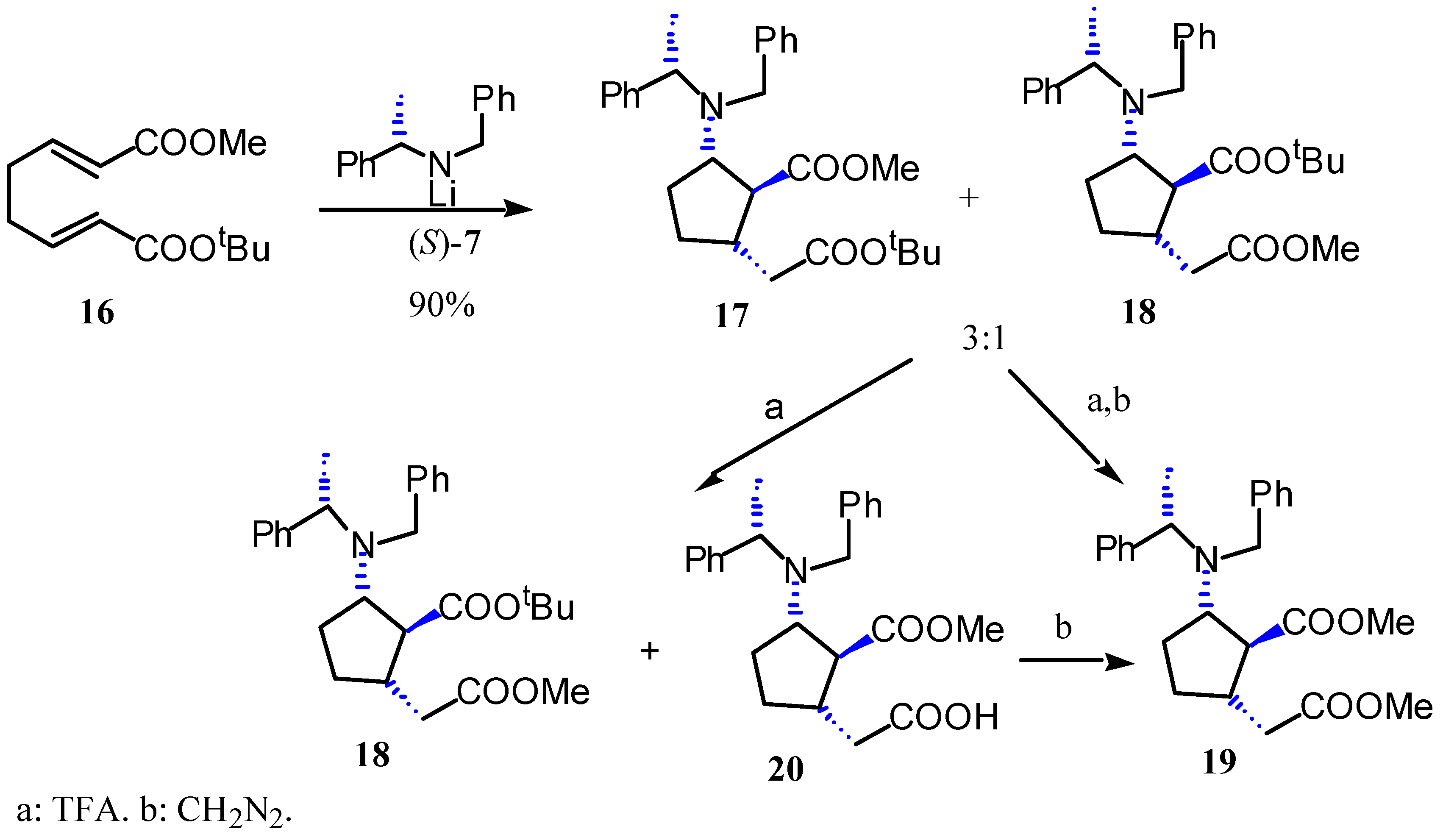
Results and Discussion
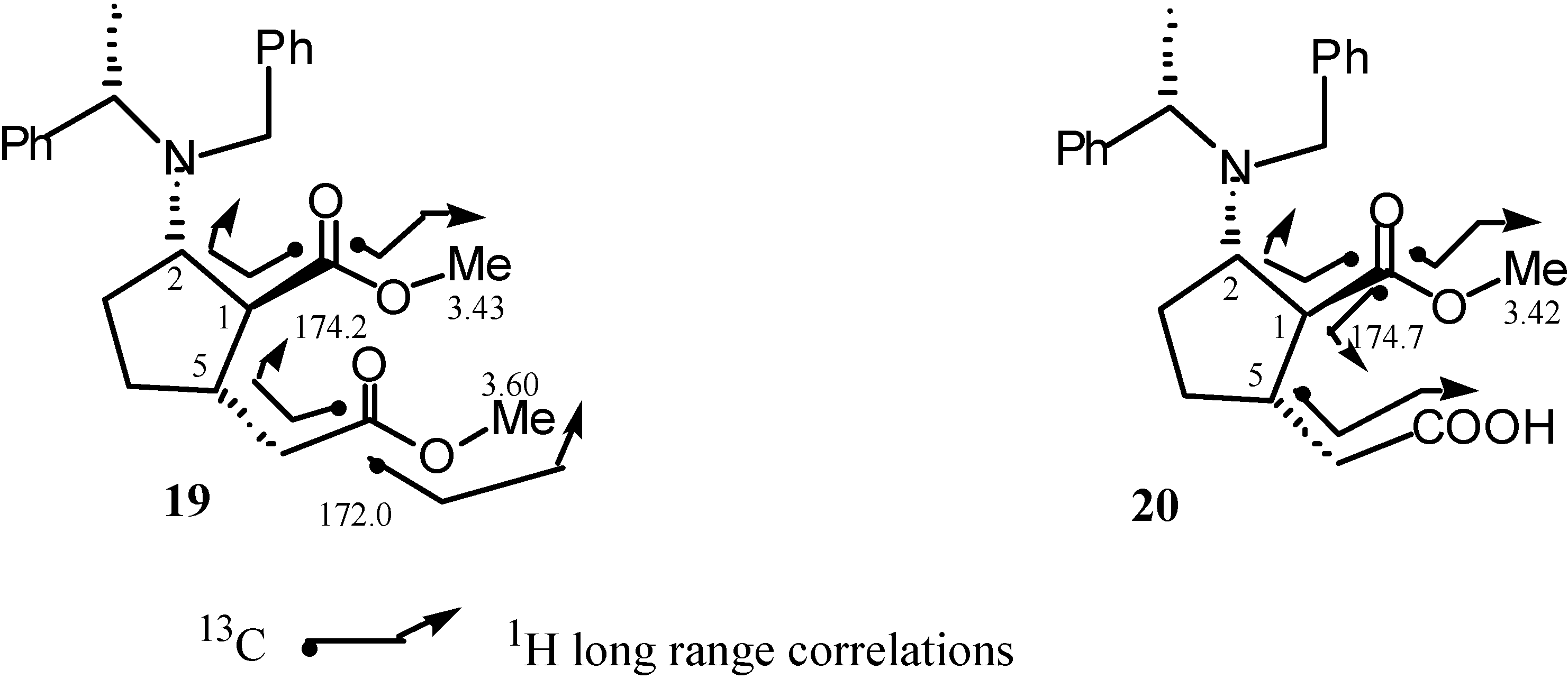
Conclusions
Acknowledgements
Experimental
General
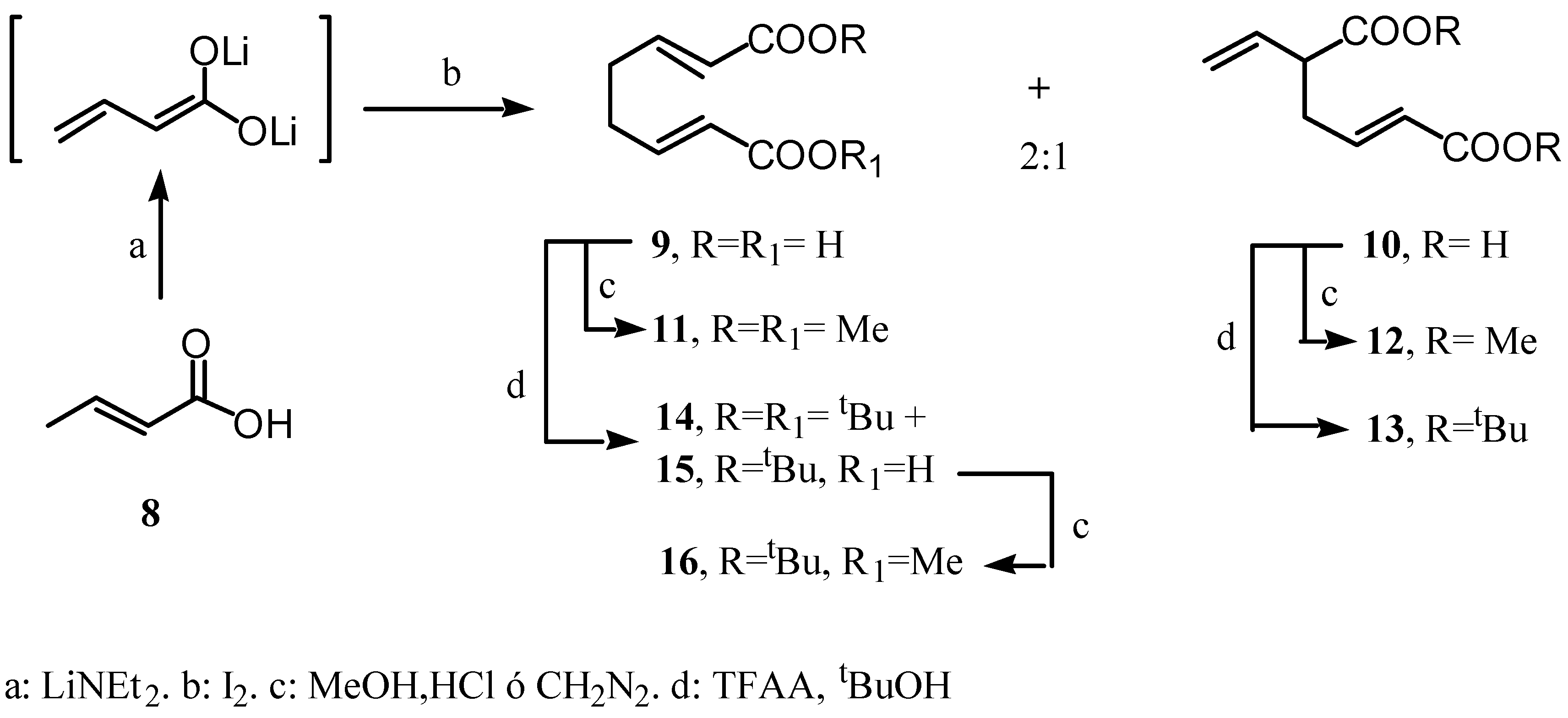
Preparation of (E)-di-tert-butyl-5-vinylhex-2-enedioate (13).
Preparation of di-tert-butyl (E,E)-octa-2,6-diendioate and tert-butyl and hydrogen (E,E)-octa-2,6-diendioates 14 and 15.
Preparation of tert-butyl methyl (E,E)-octa-2,6-diendioate (16).
Preparation of methyl (1S,2S,5S,αS)-2-N-benzyl-N-α-methylbenzylamino-5-tert-butoxycarbonyl-methylcyclopentane-1-carboxylate (17) and tert-butyl (1S,2S,5S,αS)-2-N-benzyl-N-α-methylbenzyl-amino-5-methoxycarbonylmethyl-cyclopentane-1-carboxylate (18).
Preparation of methyl (1S,2S,5S,αS)-2-N-benzyl-N-α-methylbenzylamino-5-methoxycarbonylmethyl-cyclopentane-1-carboxylate (19).
Preparation of methyl (1S,2S,5S,αS)-2-N-benzyl-N-α-methylbenzylamino-5-carboxymethylcyclo-pentane-1-carboxylate (20).
Preparation of (1S,2S,5S,αS)-2-N-benzyl-N-α-methylbenzylamino-5-methoxycarbonylmethylcyclo-pentane- 1-carboxylic acid (21).
References
- Martinek, T.A.; Táth, G.K.; Vass, E.; Hollósi, M.; Fülop, F. cis-2-Aminocyclopentanecarboxylic acid oligomers adopt a sheetlike structure: switch form helix to nonpolar strand. Angew. Chem. Int. Ed. Engl. 2002, 41, 1718–1721. [Google Scholar] [PubMed]
- Appella, D.H.; Christianson, L.A.; Klein, D.A.; Powell, D.R.; Huang, X.; Barchi, J.J.; Gellman, S.H. Residue-based control of helix shape in beta-peptide oligomers. Nature 1997, 387, 381–384. [Google Scholar]
- Appella, D.H.; Christianson, L.A.; Klein, D.A.; Richards, M.R.; Powell, D.R.; Gellman, S.H. Synthesis and structural characterization of helix-forming beta-peptides: trans-2-aminocyclo-pentanecarboxylic acid oligomers. J. Am. Chem. Soc. 1999, 121, 7574–7581. [Google Scholar] [CrossRef]
- Barchi, J.J., Jr.; Huang, X.; Appella, D.H.; Christianson, L.A.; Durell, S.R.; Gellman, S.H. Solution conformations of helix-forming beta-amino acid homooligomers. J. Am. Chem. Soc. 2000, 122, 2711–2718. [Google Scholar]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: From structure to function. Chem. Rev. 2001, 101, 3219–3232, and references cited therein. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.A.; Wang, X.; Schmitt, M.A.; Gellman, S.H. Synthesis and 12-Helical Secondary Structure of β-Peptides Containing (2R,3R)-Aminoproline. Org. Lett. 2002, 4, 3317–3319. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.A.; Wang, X.; Lee, H.S.; Wisblum, B.; Gellman, S.H. Non-haemolytic β-amino-acid oligomers. Nature 2000, 565–565. [Google Scholar]
- Woll, M.G.; Fisk, J.D.; LePlae, P.R.; Gellman, S.H. Stereoselective synthesis of 3-substituted 2-aminocyclopentanecarboxylic acid derivatives and their incorporation into short 12-helical β-peptides that fold in water. J. Am. Chem. Soc. 2002, 124, 12447–12452. [Google Scholar] [CrossRef] [PubMed]
- Urones, J.G.; Garrido, N.M.; Díez, D.; El Hammoumi, M.M.; Dominguez, S.H.; Casaseca, J.A.; Davies, S.G.; Smith, A.D. Asymmetric synthesis of the stereoisomers of 2-amino-5-carboxymethyl-cyclopentane-1-carboxylate. Org. Biomol. Chem. 2004, 2, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, J.R.; Wostradowski, R.A. Solution Photochemistry. X. A study of the effects of double-bond geometry and of increasing double-bond separation on the photochemical reactions of acyclic nonconjugated dienes. J. Org. Chem. 1972, 37, 4317–4324. [Google Scholar] [CrossRef]
- Aurell, M.J.; Gil, S.; Mestres, R.; Parra, M.; Tortajada, A. Acerca del mecanismo radicalario del acoplamiento oxidativo de dianiones de ácidos carboxílicos. Competición entre sustitución nucleofílica y transferencia electrónica. An. Quim. 1994, 90, 457–466. [Google Scholar]
- Aurell, M.J.; Gil, S.; Parra, M.; Tortajada, A.; Mestres, R. Iodine oxidative coupling of diene and triene-diolates of unsaturated carboxylic acids. Tetrahedron 1991, 47, 1997–2004. [Google Scholar]
- Aurell, M.J.; Gil, S.; Tortajada, A.; Mestres, R. A facile synthesis of 2,6-octadienedioic and 2,4,8,10-dodecatetraenedioic acids by oxidative coupling of 2-butenoic or 2,4-hexadienoic acids. Synthesis 1990, 317–319. [Google Scholar]
- Aurell, M.J.; Gil, S.; Tortajada, A.; Mestres, R.; García-Raso, A. Silver ion oxidative coupling of diene and triene-diolates of unsaturated carboxylic acids. A facile synthesis of octa- and dodeca-dienedioic acids. Tetrahedron Lett. 1988, 29, 6181–6182. [Google Scholar] [CrossRef]
- Sample Availability: Available from the authors.
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Garrido, N.M.; El Hammoumi, M.M.; Díez, D.; García, M.; Urones, J.G. A Novel Strategy Towards the Asymmetric Synthesis of Orthogonally Funtionalised 2-N-Benzyl-N-?-methylbenzylamino- 5-carboxymethyl-cyclopentane-1-carboxylic acid. Molecules 2004, 9, 373-382. https://doi.org/10.3390/90500373
Garrido NM, El Hammoumi MM, Díez D, García M, Urones JG. A Novel Strategy Towards the Asymmetric Synthesis of Orthogonally Funtionalised 2-N-Benzyl-N-?-methylbenzylamino- 5-carboxymethyl-cyclopentane-1-carboxylic acid. Molecules. 2004; 9(5):373-382. https://doi.org/10.3390/90500373
Chicago/Turabian StyleGarrido, Narciso M., Mohamed M. El Hammoumi, David Díez, Mercedes García, and Julio G. Urones. 2004. "A Novel Strategy Towards the Asymmetric Synthesis of Orthogonally Funtionalised 2-N-Benzyl-N-?-methylbenzylamino- 5-carboxymethyl-cyclopentane-1-carboxylic acid." Molecules 9, no. 5: 373-382. https://doi.org/10.3390/90500373
APA StyleGarrido, N. M., El Hammoumi, M. M., Díez, D., García, M., & Urones, J. G. (2004). A Novel Strategy Towards the Asymmetric Synthesis of Orthogonally Funtionalised 2-N-Benzyl-N-?-methylbenzylamino- 5-carboxymethyl-cyclopentane-1-carboxylic acid. Molecules, 9(5), 373-382. https://doi.org/10.3390/90500373