Introduction
Interest in selenium containing therapeutics has grown over the last thirty years [
1]. Simple organoselenium compounds have been prepared, such as selenazolopyrimidone (
1), that show antitumor activity against mouse leukemia [
2]. (Aminoethyl)phenylselenide (
2) shows excellent antihypertensive activity and selenazine derivatives such as
3 show both antibacterial and antitumor activity [
3]. Despite this, the major therapeutic benefit that selenium currently offers appears to be in the form of dietary supplements [
4]. Selenium is an essential trace element and dietary deficiency can lead to ailments including gum disease, as well as debilitating conditions such as Keshan’s disease [
4e]. The biochemistry and pharmacology of selenium is of intense current interest. Selenium is now know to be intimately involved in the activity of enzymes such as glutathione peroxidase and thioredoxin reductase, that catalyse chemistry essential to the protection of biomolecules against oxidative stress and free radical damage [
5].
Reactive oxygen species (ROS) are a byproduct of normal aerobic metabolism [
6]. Superoxide is formed when electrons leaked from the electron transport chain react with molecular oxygen and is also the product of some enzymatic processes [
6b,
7]. Superoxide dismutase converts superoxide to hydrogen peroxide, which can, in turn, lead to the formation of hydroxyl and lipid peroxyl radicals [
6b,
7]. Hydroxyl radicals are extremely reactive and can cause damage to important biomolecules that include DNA, while lipid peroxidation has been implicated in diseases such as atherosclerosis [
6a,
8]. Reactive oxygen species do have some positive roles in biology, such as providing defense against infection [
6a,
9], however, excessive concentrations of ROS can lead oxidative stress [
6a,
9]. Nature has evolved clever methods for controlling ROS. Preventative antioxidant enzymes such as glutathione peroxidase remove ROS through redox cycling [
5], while chain breaking antioxidants such as
Vitamin E, interrupt the chain reactions responsible for the formation of radical species [
10]. It should be noted that
Vitamin E refers to a family of related compounds known as
tocopherols of which α-tocopherol (
4) is most potent [
10b].
In recent years there has been much interest in developing new, more potent antioxidants such as Ebselen (
5), that also acts as a glutathione peroxidase mimic and is due to be released for human use as a non-steroidal anti-inflammatory [
11]. Water-soluble Vitamin E analogues that have been prepared include Trolox (
6) [
12], while our group has used intramolecular homolytic substitution chemistry to prepare a selenium containing analogue
7 of Vitamin E that possesses dual mode of action [
13].
Work in our laboratories has also been directed at the development of water-soluble, dual acting, tocopherol/ebselen hybrids such as
8 and to pyridine-fused tocopherol systems such as
9. Our recent efforts towards the synthesis of the latter system are described below.
Results and Discussion
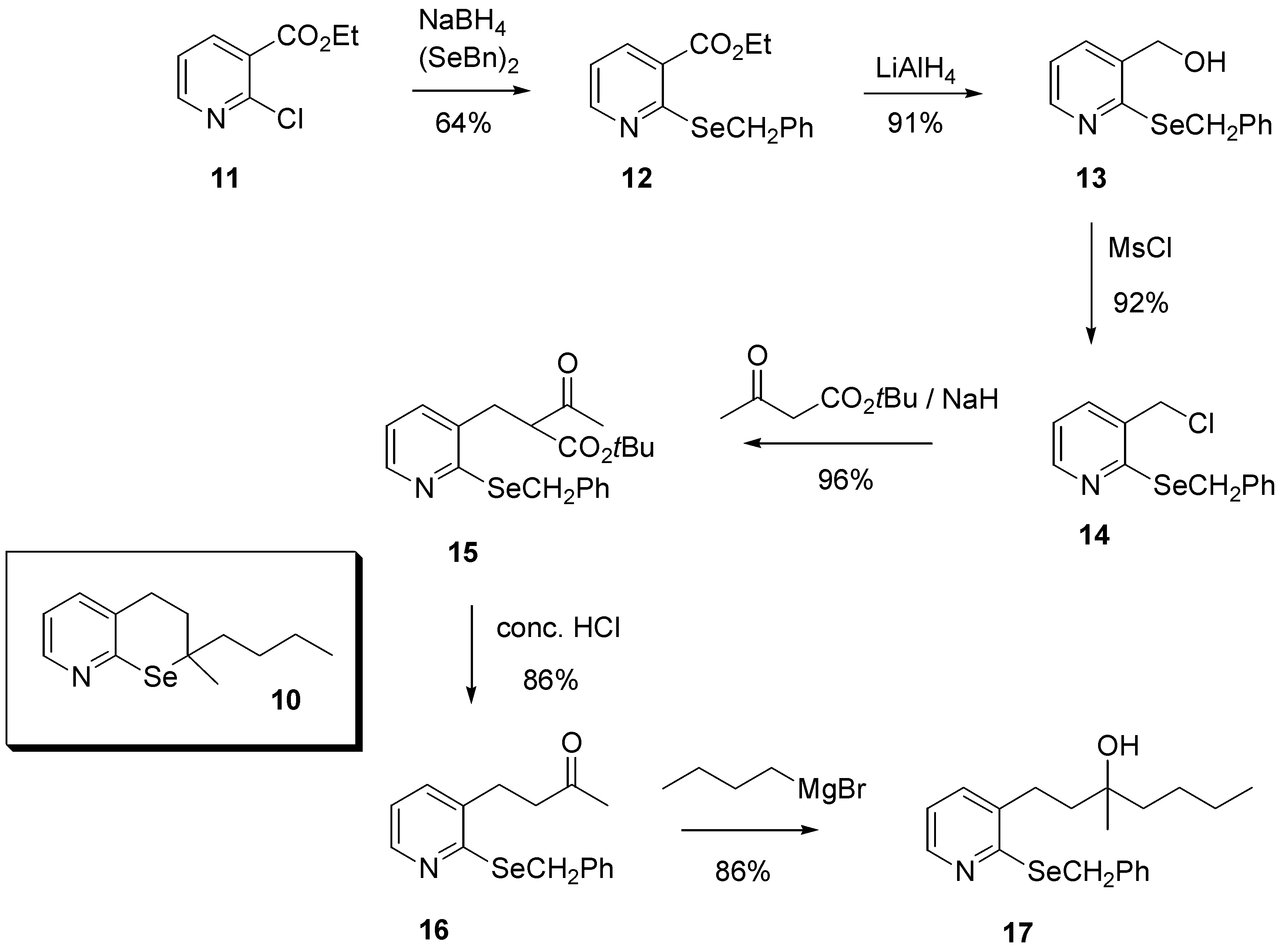
Preliminary investigations began with the attempted preparation of model compound
10 that contained many of the salient feature of structures
8 and
9. To that end ethyl 2-chloronicotinate (
11) was reacted with sodium benzylselenoate, generated
in situ by reduction of dibenzyl diselenide with sodium borohydride, to give ethyl 2-(benzylseleno)nicotinate (
12) in 64% yield (
Scheme 1).
Further reduction with lithium aluminium hydride afforded alcohol
13 in 91% yield. It is interesting to note that the alternative sequence of reactions, namely reduction of the ester
11 followed by treatment with sodium benzylselenoate proceeded only very poorly. We attribute this observation to the electrophilicity of the pyridine ring in the various substrates, specifically that the electron-withdrawing ester substituent is able to increase ring reactivity in a manner that the alcohol can not. The alcohol
13 was further reacted with methanesulfonyl chloride to give the chloride
14 in 92% yield, which was subsequently reacted with
t-butyl acetoacetate and base to give the ketoester
15 following the procedure developed by Malmström [
13]. Treatment of
15 with concentrated hydrochloric afforded the required ketone
16 in excellent overall yield. Finally, reaction of
16 with n-butylmagnesium bromide gave the desired tertiary alcohol
17 in 86% yield (
Scheme 1).
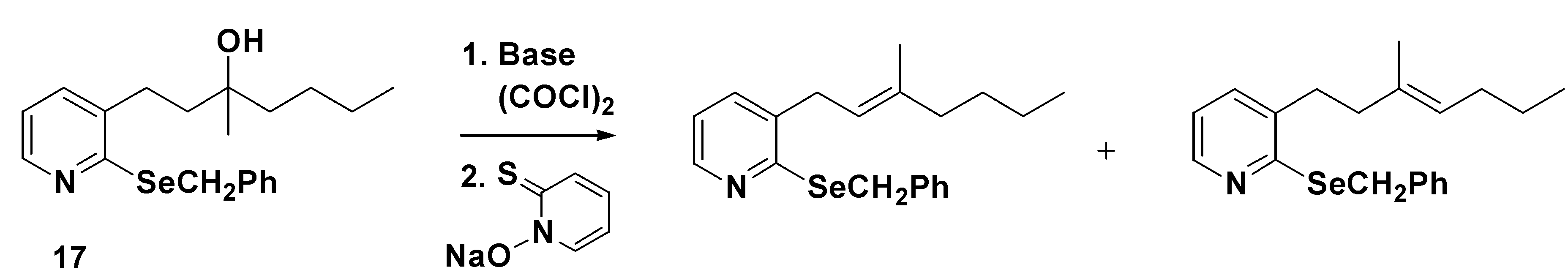
Following our previously published work, we expected that alcohol
17 could be converted into the PTOC oxalate ester, originally described by Barton and Crich as a suitable precursor for the generation of carbon-centred radicals from tertiary alcohols. To our surprise, reaction of
17 with oxalyl chloride , followed by sodium omadine
®, afforded none of the desired cyclized product
10, despite the reaction being carried out under a number of different conditions.
1H-NMR spectroscopy of the only identified product suggested strongly that elimination has occurred to afford a mixture of isomeric alkenes (
Scheme 2).
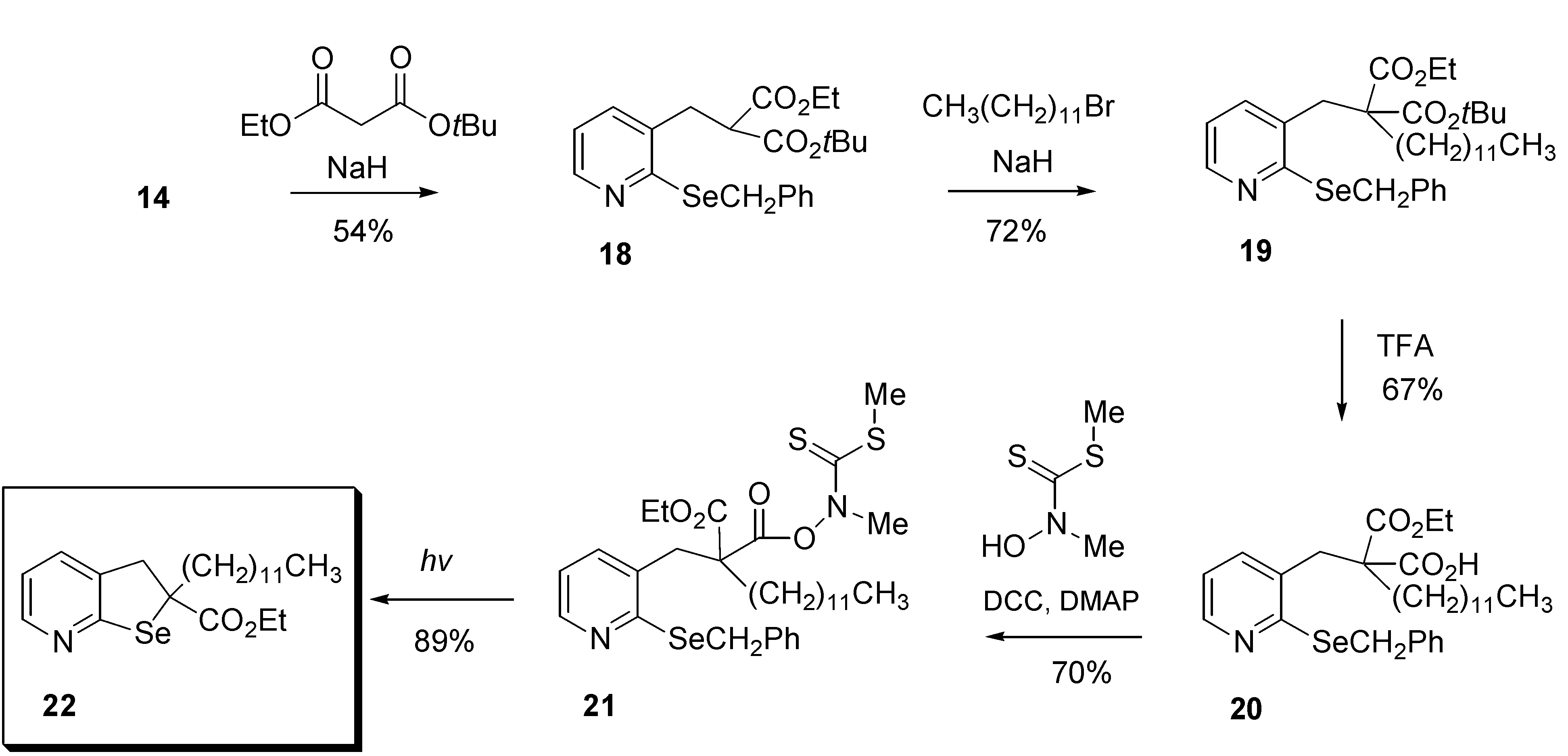
Given the problems associated with the methodology described above, it became apparent that an alternative method for the generation of the required radical was needed. Following a modification of the procedure described in
Scheme 1, chloride
14 was reacted with
t-butyl ethyl malonate to afford selenide
18 in moderate (54%) yield (
Scheme 3).
Further treatment with sodium hydride and 1-bromododecane provided diester 19 in 72% yield, which was selectively deprotected by the action of trifluoroacetic acid to afford 2-carboethoxy-2-(2-(benzylseleno)pyridin-3-yl)tridecylcarboxylic acid (20), suitable for conversion into a radical precursor.
Kim recently described a new method for generating carbon-centred radicals from thiohydroximate esters of carboxylic acids and we have generally found this precursor to be superior to those described by Barton and coworkers [
14] for reasons including stability, especially in tertiary systems [
15]. Therefore, reaction of N-methyl-hydroxydithiocarbamate and dicyclohexylcarbodiimide (DCC) with
20 afforded precursor
21 which was isolated as a yellow oil after column chromatography. Photolysis of
21 in heptane afforded the target 2-dodecyl-2-carboethoxy-2,3-dihydroselenolo[2,3-b]pyridine (
22) in 89% yield. The dihydroselenolo[2,3-b]-pyridine
22 displayed a
77Se-NMR signal at δ °547.6, characteristic of the structurally related selenium heterocycles [
16]. Having successfully demonstrated that tertiary carbon-centred radicals, such as
23 are capable of undergoing intramolecular homolytic substitution to afford dihydroselenolo[2,3-b]pyridines such as
22, we next turned our attention to the preparation of a precursor for the synthesis of a six-membered ring such as that found in the initial target compound
10.
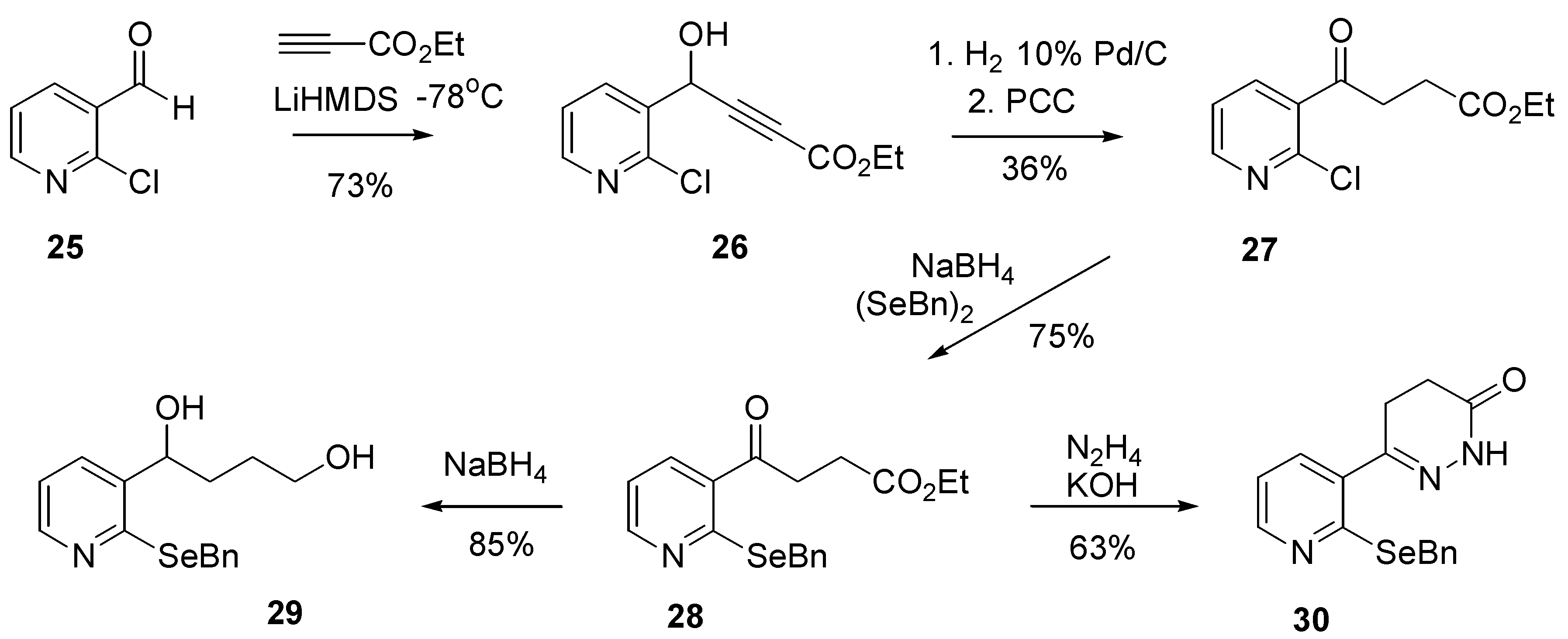
To that end, 2-chloronicotinaldehyde (
25) was added to a solution of ethyl propiolate and lithium hexamethyldisilazine in THF at –78°C (
Scheme 4).
After stirring at –78°C for 4 hours, the reaction was quenched using saturated ammonium chloride solution to give the acetylinic alcohol 26 in 73% yield after purification. It is important that the reaction mixture is quenched at low temperature, as significantly lower yields were obtained when the mixture was allowed to warm addition of the ammonium chloride solution. Hydrogenolysis of alcohol 26 followed by PCC oxidation afforded ketone 27 in poor (36%) yield. Significant amounts of by-product appeared to accompany this transformation. Despite this, ketone (27 was further reacted with sodium benzylselenoate, generated as described above, to afford ethyl 3-oxo-3-(2-(benzylseleno)pyridin-3-yl)butyrate (28) in acceptable yield.
With
28 in hand, the next task was the removal of the ketone functional group. We envisaged that Wolff-Kischner or Clemenson techniques would result in the required compound. Unfortunately, the action of zinc-mercury amalgam (Clemenson reduction) returned only
28, while reaction with alkaline hydrazine (Wolff-Kischner reduction) afforded a single product that was isolated in 63% yield and that was eventually assigned to be pyradizinone
30. The formation of
30 can be rationalized by the nucleophilic capture of the initial ketone/hydrazine adduct by the proximate ester moiety, with subsequent dehydration.
Ketone
28 proved to surprisingly difficult to reduce using standard techniques. Treatment with a large excess of sodium borohydride at room temperature resulted in no reduction, however in refluxing ethanol, clean conversion to the diol
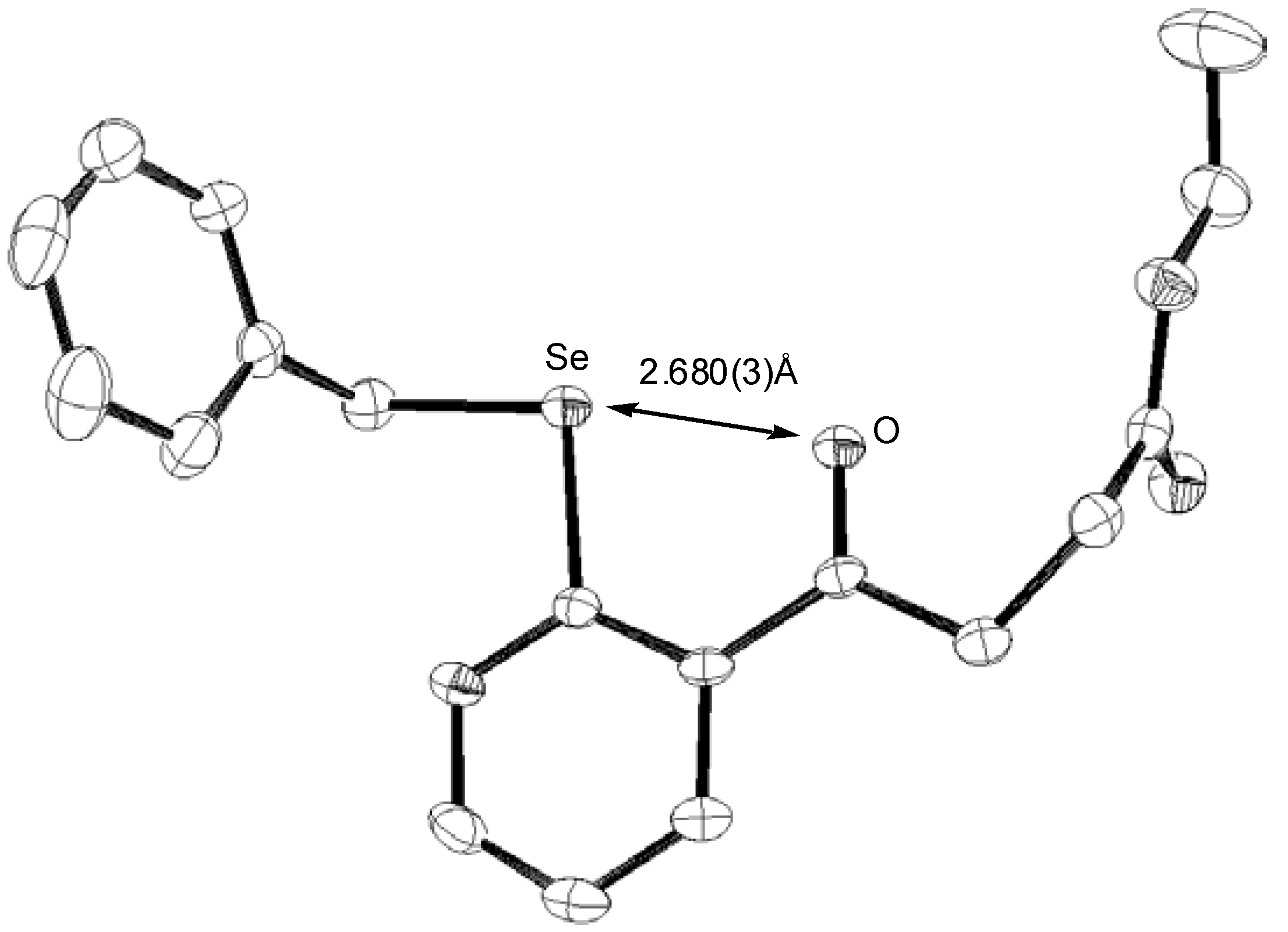
29 was observed after several hours. Single crystal x-ray analysis of
28 provided interesting structural information that may help in our understanding of the relative inertness of the ketone moiety in
28 to reduction (
Figure 1).
Figure 1.
Perspective diagram of 28
Figure 1.
Perspective diagram of 28
There appears to be a remarkably short oxygen-selenium separation in 28, which at 2.68Å is suggestive of significant contribution of resonance structure
31 to the overall bonding in
28. Indeed, the O–Se separation in
28 is, to the best of our knowledge, the shortest such interaction reported, being some 0.04Å shorter than the previous “record” of 2.72Å reported by us recently [
17].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










