Results and Discussion
Our studies focused on the extent of delocalization for the phenoxyl group’s unpaired spin density in systems with
para-phenyl,
para-vinylene and related substituents. Nishide’s success [
4e-f] in showing magnetic exchange for polyphenylenevinylenes with pendant phenoxyl radicals impelled our desire to determine how well delocalization occurs in such systems. Electron spin resonance hyperfine coupling (ESR hfc) results from such polyradicals give some information at low degrees of oxidization from precursor polyphenols to polyradicals, but the polyradicals themselves tend to have broad, featureless ESR spectra. We therefore investigated the ESR spectra for two main model systems and a set of building blocks obtained during synthesis of the main models.
Model system
1 was pursued to imitate putative polyradical
2, an all
meta-linked system. System
2 should have at best a small preference for a high spin state due to its connectivity [
1]. Although each monomer unit in
2 has an unpaired electron, with connectivity such that the number of unpaired spins nominally is equal to the degree of polymerization (n* > n° in
Figure 1)[
1b],
2 can be considered disjoint [
1c] due to the localization of molecular spin orbitals (connections only through nodal sites).
Despite their low exchange energies between unpaired electrons, such near-degenerate open-shell systems have recently become interesting due to the possibilities for spintronic [
9] strategies to “tune” their magnetic nature by application of electrical fields. We aimed to determine the stability of
1, and to find out whether delocalization was sufficient to produce an ESR-observable triplet diradical state, indicating significant exchange.
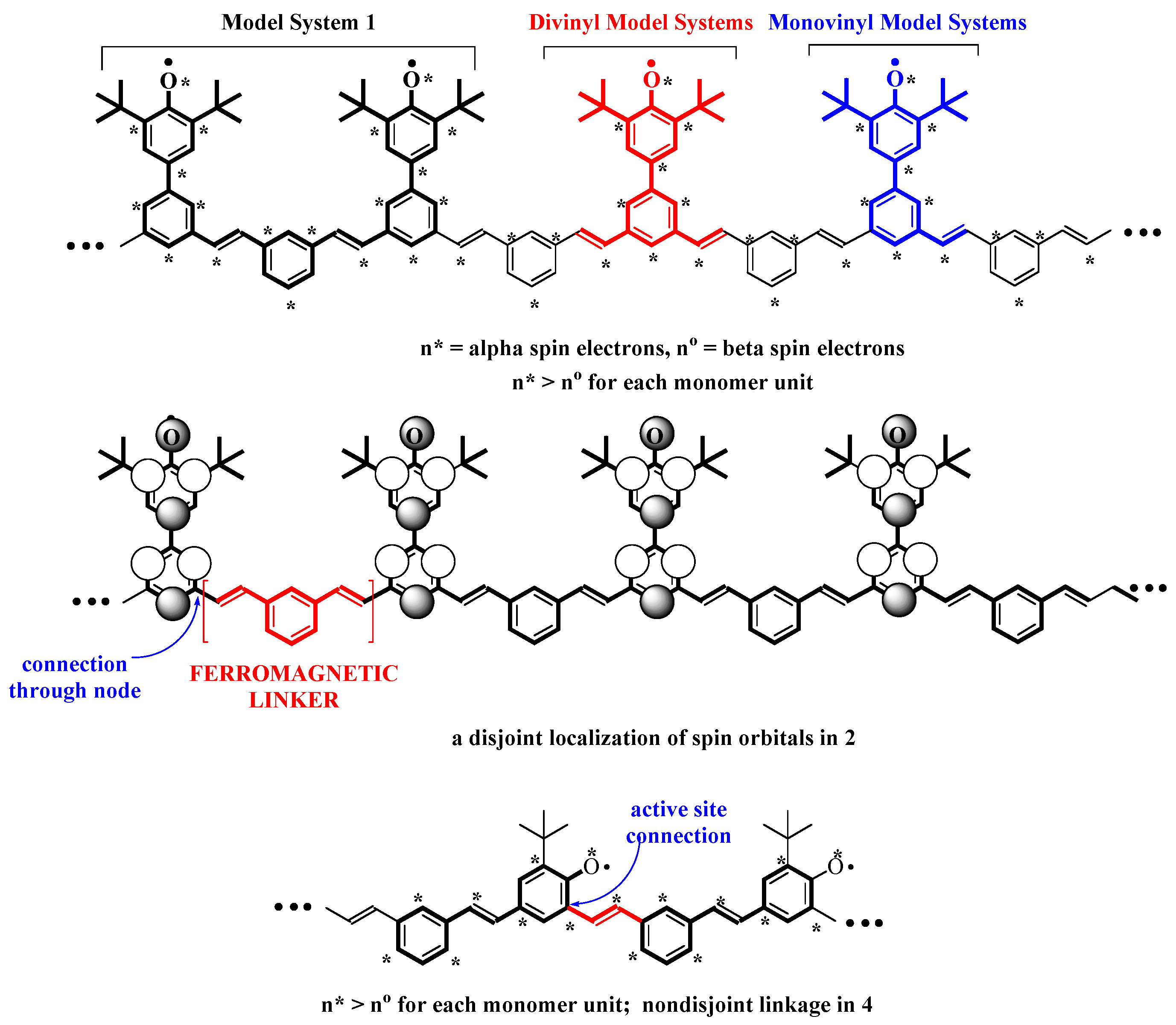
Figure 1.
Parity based analysis for polymers
2 and
4. Upper two structures show polymer
2, parsed into segments by comparison model system
1 and model systems in
Table 1 below. Lower structure shows polymer
4 parsed into stilbenic units related to
5.
Figure 1.
Parity based analysis for polymers
2 and
4. Upper two structures show polymer
2, parsed into segments by comparison model system
1 and model systems in
Table 1 below. Lower structure shows polymer
4 parsed into stilbenic units related to
5.
Model system
3 was pursued to model polyradical
4, in which a phenoxyl system would be directly incorporated into a polyphenylenevinylene chain. System
4 is expected to exhibit strong delocalization due to nondisjoint connectivity. We aimed to test its spin delocalization and stability. The related stilbenoxy system
5 has been known for some time [
10], and constitutes a useful comparison to
3, as we shall see below.
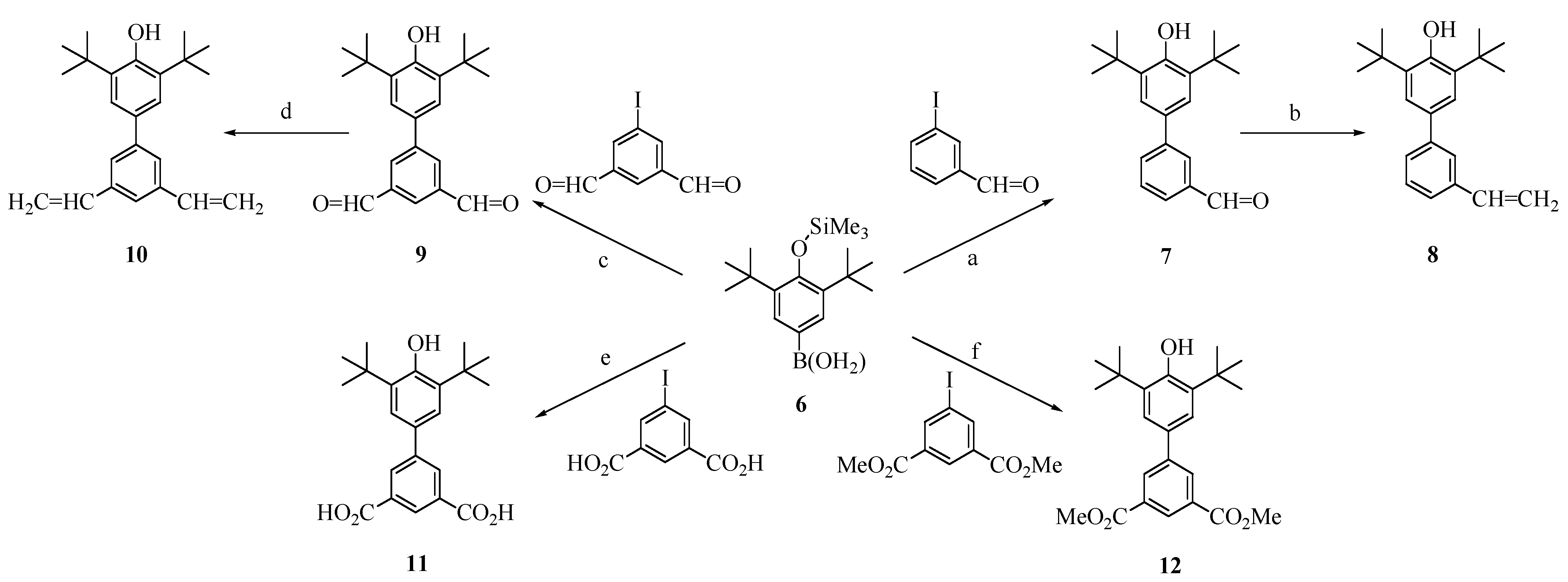
As part of investigating exchange in
1-
2, we made a number of model phenols fragments with more limited conjugation, as shown in
Figure 2. 3,5-Di-
tert-butyltrimethylsiloxyphenyl boronic acid (
6) was coupled with 3-iodobenzaldehyde by the Suzuki method to give
7 (under our reaction conditions, Suzuki couplings with
6 always gave products in which the trimethylsilyl group was cleaved). Compound
7 in turn was converted to vinyl analog
8 by a Wittig reaction. Compound
6 was also coupled with 5-iodoisophthalaldehyde by the Suzuki method, to give dialdehyde
9, which was subjected to double Wittig vinylation to yield
10. The analogous couplings with 5-iodoisophthalic acid and its dimethyl ester yielded phenols
11 and
12. These compounds constitute a set of limited-conjugation phenoxyl precursors, all having
para-phenyl substituents with
meta-linkage extension.
Figure 2.
Synthesis of fragment model segments related to polymer 2.
Figure 2.
Synthesis of fragment model segments related to polymer 2.
Conditions: (a) K2CO3, catalytic Pd(OAc)2/P(o-tolyl)3, DMF, heat, 35%; (b)CH3PPh3Br, tert-Bu-Li, THF, 80%; (c) K2CO3, catalytic Pd(OAc)2/P(o-tolyl)3, DMF, heat, 42%; (d) CH3PPh3Br, tert-Bu-Li, THF, 81%; (e) K2CO3, catalytic Pd(OAc)2/P(o-tolyl)3, acetone-water, heat, 50%; (f) K2CO3, catalytic Pd(OAc)2/P(o-tolyl)3, DMF, heat, 77%.
Figure 3 shows the synthesis of the full structure precursor to
1. 4-Bromo-2,5-di-
tert-butylphenyl acetate was reacted with 4-methoxyphenyl boronic acid using palladium catalysis (Suzuki method) to give
13, which was brominated to give
14 (even excess bromine only gave monobromination). Heck coupling of
14 with
meta-divinylbenzene gave
15 in 70% yield. Lithium aluminum hydride treatment of
15 yielded diphenol
16, the structure of which was confirmed by
1H-NMR and IR spectra. In particular, its IR spectrum showed a strong and sharp absorption for the sterically hindered phenol OH at 3600 cm
-1, and an out-of-plane
trans-HC=CH bend at 960 cm
-1. High-resolution mass spectrometry gave a molecular ion with
m/z = 750.4651, in excellent agreement with the calculated value of 750.4648 for C
52H
62O
4.
Figure 3.
Synthesis of precursor 16 to model system 1.
Figure 3.
Synthesis of precursor 16 to model system 1.
Conditions: (a) K2CO3, catalytic Pd(OAc)2/P(o-tolyl)3, DMF, heat, 64%; (b) Br2, CH2Cl2, heat, 57%; (c) Bu3N, catalytic Pd(OAc)2/P(o-tolyl)3, 1,3-divinylbenzene, DMF, heat, 44%; (d) LiAlH4, THF, 53%.
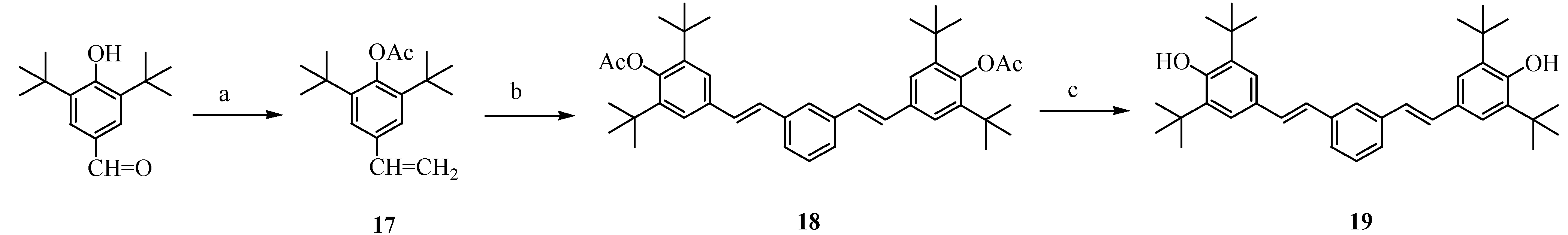
Figure 4.
Synthesis of precursor 19 to model system 3.
Figure 4.
Synthesis of precursor 19 to model system 3.
Conditions: (a) CH3PPh3Br, n-Bu-Li, THF, 60%; (b) Et3N, catalytic Pd(OAc)2/P(o-tolyl)3, 1,3-diiodobenzene, DMF, heat, 70%; (c) LiAlH4, THF, 74%.
Figure 4 shows the synthesis of the diphenolic precursor to model system
3. 3,5-Di-
tert-butyl-4-hydroxybenzaldehyde was vinylated by a Wittig reaction and acetyl-protected in a one-pot reaction to give
17, which was subjected to Heck coupling with 1,3-diiodobenzene to give
18. Subsequent deprotection yielded the diphenol
19, the structure of which was confirmed by spectroscopic and elemental analysis.
Solution phase oxidation of diphenol
16 under argon with lead dioxide in benzene or toluene immediately gave a very deep blue solution with new UV-vis bands at 319, 372, and 614 (broad) nm and the solution ESR spectrum shown in
Figure 5. The oxidation was carried out sufficient long to oxidize both phenol units in
16, based on thin layer chromatographic analysis of the consumption of di-
tert-butylphenol precursors in related oxidations (5-15 min). The spectrum was persistent for hours under inert atmosphere or
in vacuo. Analysis of the hyperfine coupling constants (hfc) was carried out using Duling’s WINSIM program [
11], showing major hfc on the phenoxyl ring and the
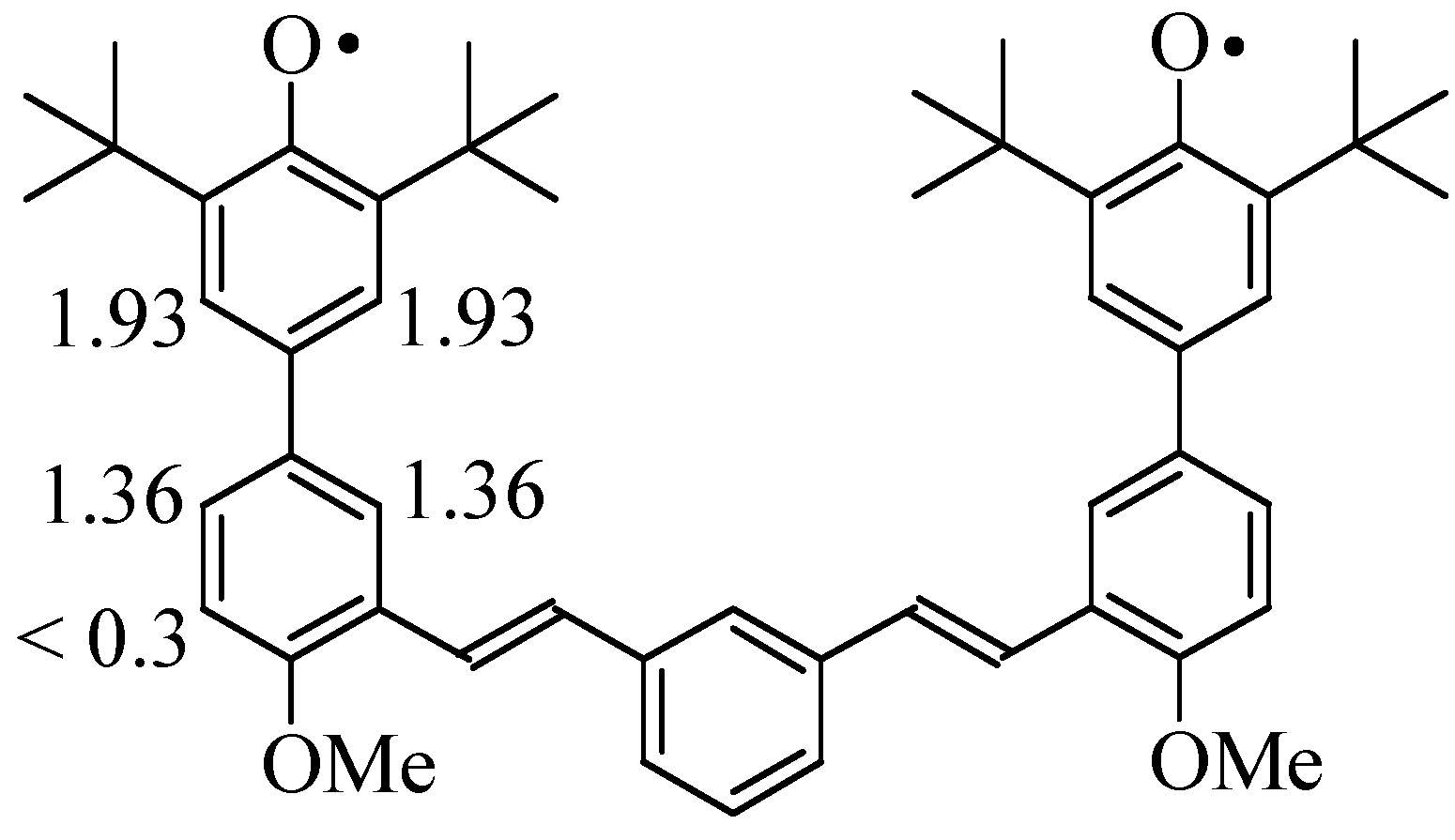
para-connected phenyl ring as expected for alternant delocalization (see
Figure 1 and
Scheme 1).
Figure 5.
Room temperature ESR spectrum derived from oxidation of 16 in toluene at 9.786 GHz
Figure 5.
Room temperature ESR spectrum derived from oxidation of 16 in toluene at 9.786 GHz
We observed no hfc indicating delocalization onto the
meta vinyl unit or beyond, despite various attempts with different solution concentrations and at various temperatures from 200-300 K. We also were unable to resolve hfc from the C-H bond
ortho to the methoxy group in the second phenyl ring, which should have a small negative spin density based on the spin distributions expected in
Figure 1. This may be due to biradical-induced broadening that is unavoidable under our conditions, or possibly to overlap of multiple small hfc that are not resolved.
Scheme 1.
Hfc assignments from
Figure 5 for spectrum assigned to
1, derived by solution oxidation of
16; hfc given in gauss.
Scheme 1.
Hfc assignments from
Figure 5 for spectrum assigned to
1, derived by solution oxidation of
16; hfc given in gauss.
If biradical
1 possessed an exchange constant J > a(H) – where a(H) are the hfc of the aromatic protons – then we would expect its line-to-line spacing to be about 50% of that observed for analogous monophenoxyl radicals [
12]. This was not observed, which would be consistent with J < a(H). A toluene solution of
1 was frozen to 77 K to see whether evidence of a discrete biradical triplet state could be detected. No fine structure attributable to ΔM
s = 1 transitions of a triplet state was observed in the g ~ 2 region, but only a single peak with linewidth of ΔH
pp = 10-11 gauss. No ΔM
s = 2 transition was observed at half-field (1600-1700 gauss region of X-band spectrum) under any conditions attempted. These observations would be consistent with a triplet state having very small zero field splitting (zfs), but if so, any exchange between the radical units of
1 must be very small. As part of the investigations of systems related to polyradical
2, we also obtained the ESR and UV-vis spectra derived by oxidation of the limited-conjugation systems with
meta-linked extension, phenols
7-
12 in
Figure 2. These were tested both as potential building blocks for future studies, and because they possess nominally conjugating substitution in the same position as the repeat units for polyradical
2.
Table 1 summarizes the UV-vis and ESR spectral data derived from brief oxidation of these precursors to radicals. In this Table a(H) are proton hyperfine constants in gauss, derived from on lineshape fitting using WINSIM [
11].
Table 1.
ESR hyperfine and UV-vis data for model phenoxyl radicals derived from phenols in
Figure 2. All spectra obtained in benzene or toluene solutions at room temperature.
Table 1.
ESR hyperfine and UV-vis data for model phenoxyl radicals derived from phenols in Figure 2. All spectra obtained in benzene or toluene solutions at room temperature.
| Compound | a(H)/gauss | UV-vis (λmax, nm) |
|---|
![Molecules 09 00725 i002]() | R = CHO | a3 = 1.80 (2 H) a5 = 0.68
a4 = 1.60 (2 H) a6 = 1.86 | 350, 482, 599 (broad)
dark green |
| R = CH=CH2 | a3 = 1.76 (2 H) a5 = 0.69
a4 = 1.70 (2 H) a6 = 1.73 | 328, 355, 507
red-purple |
![Molecules 09 00725 i003]() | R = CHO | a3 = 1.77 (2 H) a6 = 1.78
a4 = 1.65 (2 H) | 341, 458, 615(broad)
dark green |
| R = CH=CH2 | a3 = 1.68 (2 H) a6 = 1.92
a4 = 1.64 (2 H) | 276, 337, 514 (broad)
red-purple |
| R = CO2H | a3 = 1.76 (2 H)* a6 = 1.86
a4 = 1.67 (2 H) | 267, 359, 505 (broad)*
(yellow) |
| R = CO2Me | a3 = 1.69 (2 H) a6 = 1.70
a4 = 1.69 (2 H) | 347, 475, 615 (broad)
(blue) |
All radicals exhibited highly colored solutions that were persistent for hours if exposure to air was limited. All show significant hfc in both rings, showing delocalization from the phenoxyl onto the phenyl ring – however, in no case did we obtain resolvable hfc from the linking substituent in the
meta-position (R in the table). The sum of all available information for both
1 and the radicals derived from
7-
12 shows no more than 2-3% spin delocalization onto any π-orbital bearing carbon at the
meta-position (R = vinyl, carbonyl carbons in
Table 1), if our resolution is at least a conservative 0.5 G, and the McConnell relationship [
13] holds such that a(H) = (-22 G) × ρ(πC), where ρ(πC) = π-spin density of carbon to which an aryl C-H is attached with an hfc of a(H).
Although the ESR evidence shows neither significant delocalization of unpaired spin onto the
meta-linked positions of
7-
12, nor major variation with substituent R (
Table 1), the UV-vis spectra show a qualitative trend. The vinyl-substituted phenoxyl radicals derived from
8 and
10 show substantially blue-shifted long-wavelength bands relative to the carbonyl-substituted variants. The diacid system (R=CO
2H,
Table 1) is yellow in benzene or toluene, but greenish in DMSO, so this phenoxyl may not fit the trend well due to its known hydrogen bonding in nonpolar solvents [
14]. The trend may be due to differing charge-transfer character of the long wavelength band in the systems with the more electron-poor ring, namely those with the carbonyl substituents.In an effort to test a nondisjoint system where greater delocalization is expected, diphenol
19 was stirred with PbO
2 in benzene for 1-5 min to give deep yellow solutions. The color change was accompanied by appearance of a strong band in the UV-vis spectrum at 309 nm and quite weak bands at 457 and 488 nm. Oxidation of the precursor phenol groups was confirmed by disappearance of the OH stretching absorption at 3600 cm
-1 in the IR spectrum of a film of oxidized product. The ESR spectrum exhibited the multi-line pattern shown in
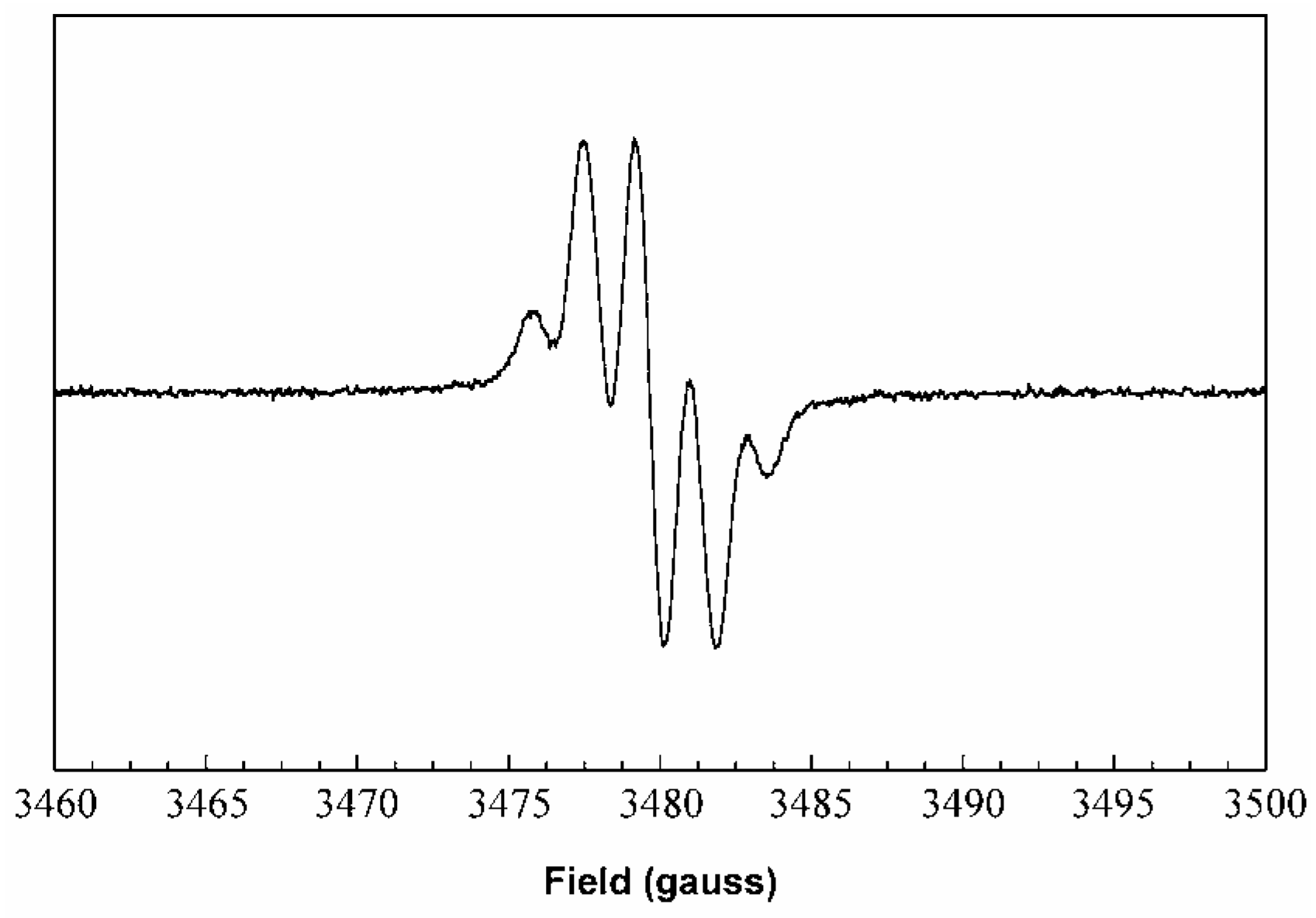
Figure 7, which shows an excellent fit (correlation coeff = 0.993) to the following hfc: a(H) = 6.37 G (1 H), 2.93 (1 H), 1.69-1.79 (2 H), 1.40-1.48 (3 H), 0.53 G (1 H). The spectrum did not vary with the length of oxidation over 1-5 min, and persists for hours in the absence of air.
Figure 7.
Room temperature ESR spectrum derived from oxidation of
19. Experimental spectrum in toluene at 9.751 GHz (black); simulation (red) with WINSIM [
11] using a(H) = 6.37 (1 H), 2.93 (1 H), 1.69 (1 H), 1.59 (1 H), 1.48 (1 H), 1.47 (1 H), 1.40 (1 H), 0.53 G (1 H).
Figure 7.
Room temperature ESR spectrum derived from oxidation of
19. Experimental spectrum in toluene at 9.751 GHz (black); simulation (red) with WINSIM [
11] using a(H) = 6.37 (1 H), 2.93 (1 H), 1.69 (1 H), 1.59 (1 H), 1.48 (1 H), 1.47 (1 H), 1.40 (1 H), 0.53 G (1 H).
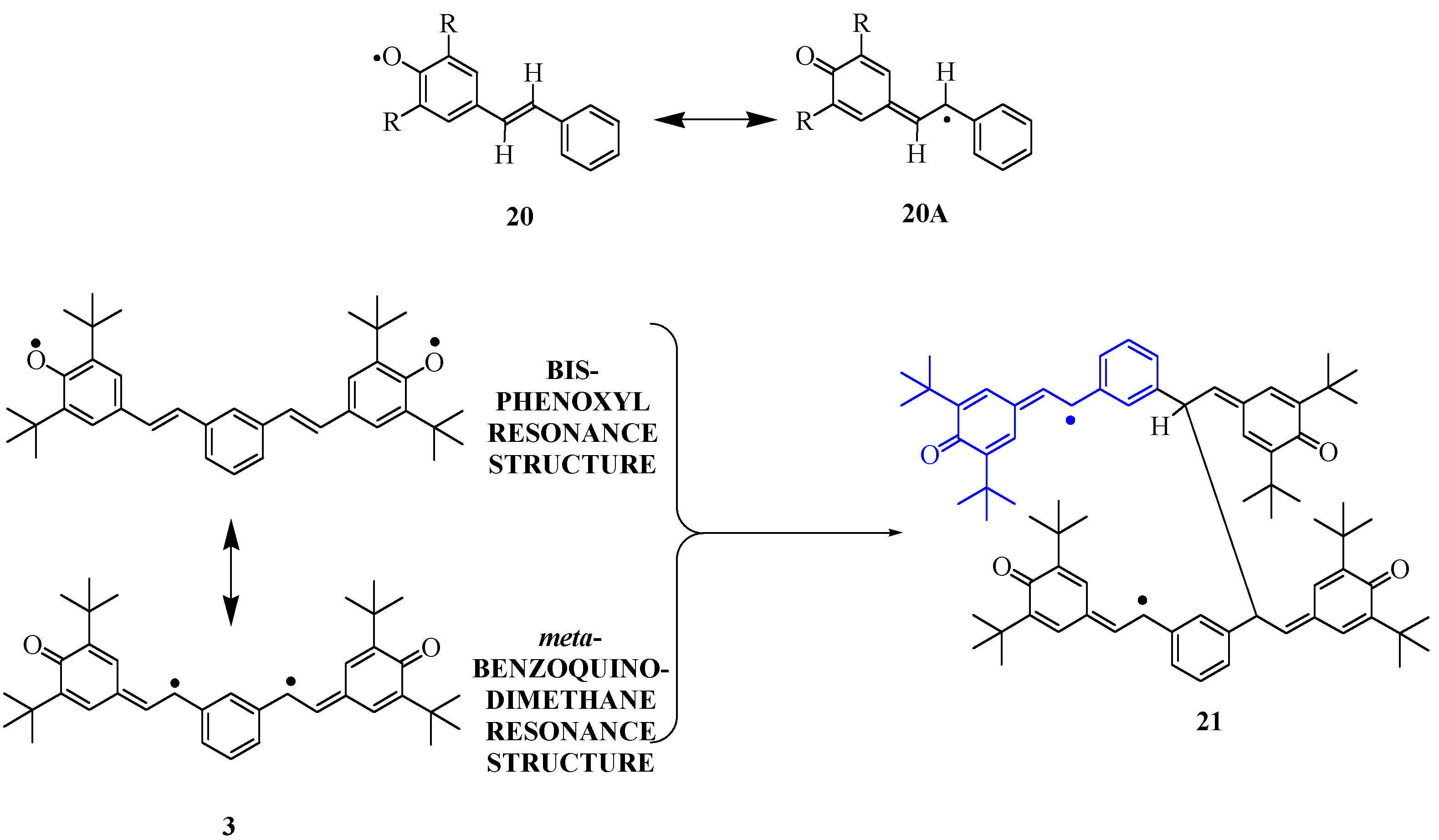
The spectrum from
19 exhibits an unusually large hfc of 6.4 G for one proton, requiring that the proton be attached at a position with a large spin population (nearly 30% based on the McConnell model described above). The hfc of the
meta-hydrogen atoms on a
tert-butylated phenoxyl ring are typically 1-3 G. We therefore assigned the large hfc to a stilbenic olefin CH group, due to resonance structure
20A (R=
tert-Bu,
Scheme 2). This is consistent with computations for 4-stilbenoxy model
20 (R=H) at the UB3LYP/EPR-II // UB3LYP/6-31G* level using Gaussian 98 [
15]. The computations predict hyperfine couplings in good accord with those recently [
16] reported for radical
20 (R=
tert-Bu); the report gives one proton hfc of 6.39 G. Clearly,
20 and related systems are so delocalized that their major spin density site is not even found in the phenoxyl ring, but instead on the olefinic group. This tendency is strengthened by the significant C=O bonding character in resonance structure
20A.
The 4-stilbenoxyl
20 (R=
tert-Bu) dimerizes readily in the solid state [
10], although it shows a persistent solution ESR spectrum. We presume that the analogous system
3 is similarly reactive to form
21 or related species, explaining our inability to observe a triplet state spectrum for
3 despite expected strong exchange in this nondisjoint connectivity.
Scheme 2.
Resonance structures of 4-stilbenoxyl radicals, and a consequent, likely dimerization pathway for diradical 3.
Scheme 2.
Resonance structures of 4-stilbenoxyl radicals, and a consequent, likely dimerization pathway for diradical 3.
Given the strong delocalization of the phenoxyl spin density on the stilbenic olefin units, system
3 is probably more like a
meta-benzoquinodimethane than a bis-phenoxyl (
Scheme 2). The position of highest spin density in
3 is not sterically protected, hence the diradical is so reactive that it is not observed when generated in solution. Apropos, one must parenthetically note that organic polyradical synthesis for magnetic materials involves a delicate balance between spin delocalization that is desirable because it promotes stronger exchange interaction between spin sites, and excessive spin delocalization that leads to undesirable radical couplings and formation of defects in the polyradical. Assuming that
3 dimerizes as shown in
Scheme 2, the resulting product should show hfc very much like those described for
20 (R=
tert-Bu), but lacking one of the 0.5-0.6 G hyperfine couplings. This is exactly what is observed. Based on this analysis, we assigned the hfc as shown in
Scheme 3 for
20 (R=
tert-Bu) and the putative dimer of biradical
3, structure
21. The ability of
21 to survive further reaction to completely ESR-silent products is consistent with the similar persistence of
20 (R=
tert-Bu) in solution. Since we do not see any evidence of biradical exchange in the spectrum from the oxidation of
19, we presume that interruption of the conjugation pathway precludes exchange if two radical sites in
21 are simultaneously present.
Scheme 3.
Hfc assignments for model systems and
21. Hfc computed by UB3LYP/EPR-II method for
20(H) are red. See [
10] and [
16] for literature descriptions of ESR for
20 (
t-Bu). See
Scheme 2 above for full, proposed structure of
21.
Scheme 3.
Hfc assignments for model systems and
21. Hfc computed by UB3LYP/EPR-II method for
20(H) are red. See [
10] and [
16] for literature descriptions of ESR for
20 (
t-Bu). See
Scheme 2 above for full, proposed structure of
21.
Experimental
General
All 1H-NMR spectra were obtained using either an IBM Instruments NR-80A (80 MHz) or a Bruker AC-200 (200 MHz) instrument. All spectra were recorded using chloroform-d6 or acetone-d6 and were calibrated using tetramethylsilane as internal standard. All IR spectra were recorded on a Perkin Elmer PE-1420 spectrometer or a Midac M-2000 FTIR with computer interface. UV-Vis spectra were recorded on a Shimadzu UV-260 double beam spectrometer. Melting points were obtained on Electrothermal IA6404 melting point apparatus, and are reported without correction. ESR spectra were obtained on a Bruker ESP-300 X-band (~ 9 GHz) spectrometer with standard computer interface and cooled nitrogen-gas variable temperature attachment. Solutions of stable radicals for ESR determination were subjected to three cycles of freeze-vacuum-thaw in order to remove oxygen, using quartz 4 mm O.D. tubes with a stopcock on top. All the reactions were run under an atmosphere of argon unless otherwise stated. All chemicals were purchased from Aldrich Chemical Company except where noted. All solvents were purchased from Fisher Scientific except where noted and were used without further purification except where noted. Dry tetrahydrofuran (THF) was distilled freshly under argon from sodium-benzophenone after pre-drying over lithium aluminum hydride. Anhydrous diethyl ether was either used directly from a freshly opened container or was distilled from potassium or sodium-benzophenone. Dry methylene chloride was distilled from calcium chloride. Dry triethylamine was distilled from phosphorus pentoxide. Dry dimethylformamide (DMF) was either purchased from Aldrich Chemical Company in a SureSeal™ bottle or freshly distilled from phosphorus pentoxide.
Dimethyl 5-iodoisophthalate
5-Iodoisophthalic acid (0.7 g, 2.4 mmol) was placed in a round bottom flask together with methanol (20 mL) and conc sulfuric acid (0.5 mL), and the mixture was refluxed overnight. Most of the methanol was removed under reduced pressure and the residue was dissolved in ethyl acetate. The organic layer was washed with water until the wash was neutral and dried over magnesium sulfate. A brown solid obtained after removal of the organic solvent was found to be a mixture. Silica gel column chromatography with 20/80 ethyl acetate/hexane separated the desired compound as a white crystalline solid (0.5 g, 65 %), mp 103-104.5 °C (lit. [
17] 100-102 °C);
1H-NMR (200 MHz, CDCl
3) δ 8.64-8.63 (t, 1H, J=1.46 Hz), 8.56-8.55 (d, 2H, J=1.44 Hz), 3.95 (s, 6H); MS-EI (m/z): calc’d for C
10H
9IO
4: 320; found: 320.
5-Iodoisophthaldehyde
Into a suspension of lithium aluminum hydride (0.62 g, 16.3 mmol) in dry diethyl ether (20 mL) cooled in an ice bath was added via a syringe a solution of dimethyl 5-iodoisophthalate (1.74 g, 5.44 mmol) in diethyl ether (5 mL). After the addition, the reaction mixture was stirred at room temperature for two more hours. The mixture was then poured into ice and extracted with ethyl acetate. The organic layer was washed with brine and dried over magnesium sulfate. The product,
1,3-dihydroxymethyl-5-iodobenzene was obtained as a yellowish powder (0.94 g, 65 %) after removal of the solvent, and was used without further purification for oxidative conversion to 5-isophthalaldehyde.
1H-NMR (200 MHz, CDCl
3) δ 7.62 (m, 2H), 7.34 (m, 1H), 4.69 (s, 4H) [
18]. A mixture of the dihydroxy product (0.8 g, 3.03 mmol) from the previous step was heated under reflux for 2 h with pyridinium chlorochromate (1.95 g, 9.09 mmol) in dichloromethane (20 mL). TLC showed disappearance of the starting material and presence of a major spot with higher R
f. The reaction mixture was adsorbed on silica gel and eluted with 20/80 ethyl acetate/hexane. The desired compound was obtained after removal of solvent as a white crystalline solid with mp 117-119 °C (0.6 g, 76 %);
1H-NMR (200 MHz, CDCl
3) δ 10.03 (s, 2H), 8.46-8.45 (d, 2H, J=1.44 Hz), 8.34-8.32 (t, 1H, J=1.44 Hz); IR (neat, cm
-1): 1690.
3,5-Di-tert-butyl-4-trimethylsiloxyphenyl boronic acid (6)
n-Butyllithium (3.7 mL of a 1.6 M solution in hexane, 5.9 mmol) was added into dry THF (10 mL) and the resulting yellow solution was cooled to -78
oC in a Dry Ice-acetone bath. A solution of 4‑bromo-2,6-di-
tert-butyltrimethylsiloxyphenol [
19] (2.0 g, 5.6 mmol) in dry THF (15 mL) was added in via a cannula and the mixture was stirred at -78 ºC for 20 min. Triisopropylborate (1.14 g, 6.1 mmol) was added via a syringe and stirring was continued for twenty more minutes. The mixture was then warmed up to room temperature, poured into a solution of hydrochloric acid (2 mL hydrochloric acid in 100 mL water), stirred for 2 h and extracted with ethyl acetate. The organic layer was washed with water and dried over magnesium sulfate. A yellowish powder was obtained after removal of the organic solvent. Recrystallization from 60/40 ethyl acetate/hexane gave a white crystalline solid (1.33 g, 74 %) with mp 230 °C (decomp.) (lit. mp 235-237 °C for the cyclic boroxin trimer of
6 [
20]).
1H-NMR (200 MHz, CDCl
3) δ 8.16 (s, 2H), 1.49 (s, 18H), 0.45 (s, 9H).
3,5-Di-tert-butyl-4-hydroxy-3’-vinylbiphenyl (8)
Methyltriphenylphosphonium bromide (0.26 g, 0.71 mmol) was suspended in THF (15 mL) and cooled in an ice bath. tert-Butyllithium (0.46 mL of a 1.7 M solution in hexane, 0.78 mmol) was added via a syringe giving a bright yellow solution. A solution of 7 (0.1 g, 0.32 mmol) in THF (5 mL) was added dropwise via a pressure-equalizing addition funnel. The color of the reaction turned to brown after the addition. The mixture was extracted with ethyl acetate and the organic layer was washed with water and dried over magnesium sulfate. The residue after removal of the organic solvent was applied to a silica gel column and eluted with 20/80 ethyl acetate/hexane. The desired compound was obtained as a yellow oil that crystallized into the yellow solid 8 (0.08 g, 80 %), with mp 72-73.5 °C. 1H-NMR (200 MHz, CDCl3) δ 7.55-7.26 (m, 6H), 6.86-6.72 (dd, 1H, J=17.34 Hz, 10.82 Hz), 5.85-5.76 (dd, 1H, J=17.68 Hz, 1.08 Hz), 5.31-5.26 (dd, 1H, J=10.84 Hz, 0.74 Hz), 5.28 (s, 1H), 1.50 (s, 18H). IR (neat, cm-1): 3636, 2957, 989, 906. UV-Vis (benzene, λmax, nm): 279.0. Elemental analysis: calc’d for C22H28O: C 85.71, H 9.09; found: C 84.86, H 9.24.
3,5-Di-tert-butyl-3’,5’-divinyl-4-hydroxybiphenyl (10)
Into a suspension of methyltriphenylphosphonium bromide (0.35 g, 0.98 mmol) in THF (15 mL) cooled in an ice bath was added tert-butyllithium (0.63 mL of a 1.7 M solution in hexane, 1.07 mmol). A bright yellow color formed. Compound 9 (0.1 g, 2.96 mmol) in THF (2 mL) was added slowly via a syringe giving a pale yellow color. The mixture was stirred at room temperature for two more hours. TLC showed the main product was blue under short wave ultraviolet light and had a high Rf. The mixture was then poured into water, acidified with hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with water, dried over magnesium sulfate, and evaporated. The residue was chromatographed (silica gel, 20/80 ethyl acetate /hexane) to give the product as a yellow crystalline solid (0.80 g, 81 %), mp 92-94 °C; 1H-NMR (200 MHz, CDCl3) δ 7.44-7.38 (m, 5H), 6.86-6.72 (dd, 2H, J=17.32 Hz, 10.82 hH), 5.87-5.78 (dd, 2H, J=17.68 Hz), 5.33-5.27 (dd, 2H, J=0.72 Hz), 5.29 (s, 1H), 1.50 (s, 18H); IR (neat, cm-1): 3631, 2957, 988, 907; UV-Vis (benzene, λmax, nm): 278.0; Elemental analysis: calc’d for C24H30O: C 86.22, H 8.98; found: C 85.99, H 9.00.
3,5-Di-tert-butyl-4-hydroxybiphenyl-3’,5’-dicarboxylic acid (11)
Compound
6 (0.13 g, 0.403 mmol), 5-iodoisophthalic acid (0.11 g, 0.38 mmol), palladium acetate (10 mg, 5 mol-%) and potassium carbonate (1.0 g, 7.25 mmol) were added to a three-neck round bottom flask that was flushed with argon. A 1:1 mixture of acetone-water (40 mL) was added by a syringe and the mixture was heated at reflux under argon for 2 h. A black green mixture resulted. Acetone was removed under reduced pressure and the residue was extracted three times with ethyl ether and the organic layer was discarded. The aqueous layer was acidified with hydrochloric acid and then extracted with ethyl ether. The ether layer was washed with water and dried over magnesium sulfate and a yellowish solid (70 mg, 50%) was obtained after removal of solvent, mp 290 °C (d);
1H‑NMR (200 MHz, acetone-
d6) δ 8.61-8.60 (t, 1H, J=1.6 Hz), 8.43-8.42 (d, 2H, J=1.6 Hz), 7.52 (s, 2H), 1.52 (s, 18H); IR (KBr, cm
-1): 3630, 3449, 2922, 1702; UV-Vis (benzene, λ
max, nm): 290. A small amount of the sample was dissolved in acetone and the solvent was allowed to evaporate slowly to yield single crystals. X-ray diffraction analysis carried out by Prof. Clifford George of the Laboratory for the Structure of Matter at the Naval Research Laboratory. The structure was found to be the product in 1:1 ratio with acetone [
14]. Elemental analysis: calc’d for C
22H
26O
5°C
3H
6O: C 70.09, H 7.48; found: C 70.00, H 7.59. Details of the crystal structure are available through the Cambridge Crystallographic Databank (CCDC Deposition # 137194).
Dimethyl 3,5-di-tert-butyl-4-hydroxybiphenyl-3’,5’-dicarboxylate (12)
3,5-Di-tert-butyl-4-trimethylsiloxyphenyl boronic acid (6, 0.51 g, 1.56 mmol), dimethyl 5‑iodoisophthalate (0.5 g, 1.56 mmol), palladium acetate (18 mg, 5 mol %), tri-o-tolylphosphine (95 mg, 20 mol %) and potassium carbonate (1.1 g, 8.0 mmol) were added to a three-neck round bottom flask that was flushed with argon for 20 min. Dry DMF (30 mL) was added, and the mixture was heated at 70 °C for 1 h. A dark-brown color resulted. The mixture was poured into water and extracted with ethyl acetate. The organic layer was washed thoroughly with water and brine to remove DMF and dried over magnesium sulfate. The residue after removal of solvent was applied to a silica gel column and the main product was isolated as a yellowish solid (0.46 g, 77%), mp 160-162 °C. 1H-NMR(200 MHz, acetone-d6) δ 8.61-8.59 (t, 1H, J=2.0 Hz), 8.38-8.37 (d, 2H, J=2.0 Hz), 7.41 (s, 2H), 5.36 (s, 1H), 3.98 (s, 6H), 1.51 (s, 18H). UV-Vis (benzene, λmax, nm): 284.2. Elemental analysis: calc’d for C24H30O5: C 72.36, H 7.54; found: C 72.60, H 7.69.
Oxidation of 7
A small amount of 7 (ca. 5 mg) was dissolved in benzene (1 mL) and lead dioxide was added. A dark green solution resulted, which was used for ESR and UV-Vis determination. UV-Vis (benzene, λmax, nm): 350, 482, 599; ESR (9.651 GHz, benzene, gauss): a(H) = 1.80 (2H), 1.60 (2H), 1.86, 0.68.
Oxidation of 8
A small amount of 8 (ca. 5 mg) was dissolved in benzene (1 mL) under argon and lead dioxide powder. The solution color changed to red-purple immediately and this solution was used for spectral analysis. UV-Vis (benzene, λmax, nm): 328, 355, 507; ESR (9.670 GHz, benzene, gauss): a(H) = 1.76 (2H), 1.70 (2H), 1.73, 0.64.
Oxidation of 9
A small amount of 9 (ca. 5 mg) was dissolved in benzene (1 mL) and excess lead dioxide powder was added. A dark green solution resulted, which was used for ESR and UV-Vis determination. UV-Vis (benzene, λmax, nm): 341, 458, 615; ESR (9.652 GHz, benzene, gauss): a(H) = 1.77 (2H), 1.65 (2H), 1.78.
Oxidation of 10
A small amount of 10 (ca.5 mg) was dissolved in benzene (1 mL) under argon and excess lead dioxide powder was added. A red purple solution was formed at once. The solution was used directly for ESR. UV-Vis (benzene, λmax, nm): 276, 337, 514; ESR (9.664 GHz, benzene, gauss): a(H) = 1.68 (2H), 1.64 (2H), 1.92.
Oxidation of 11
To a dilute benzene solution of the radical precursor 11, prepared by gentle warming and recooling, was added lead dioxide powder. The solution color changed to greenish yellow. The solution was used at once for spectral investigation. UV-Vis (benzene, λmax, nm): 267, 358, 505; ESR (9.795 GHz, benzene, gauss): a(H) = 1.83 G (2H), 1.72 G (2H), 1.62.
Oxdiation of 12
A small amount of 12 (ca. 5 mg) was dissolved in benzene (1 mL) and lead dioxide powder was added. A blue color formed immediately. The resulting blue solution was used for ESR and UV-Vis determinations. UV-Vis (benzene, λmax, nm): 347, 475, 615; ESR (9.800 GHz, benzene, gauss): a(H) = 1.70 (2H), 1.69 (2H), 1.69.
4-Acetoxy-3,5-di-tert-butyl-4’-methoxybiphenyl (13)
4-Bromo-2,6-di-
tert-butylphenyl acetate [
21] (2.0 g, 6 mmol), 4-methoxyphenyl boronic acid (
8) (0.93 g, 6 mmol), palladium acetate (68 mg, 5 mol-%), tri-
o-tolyl-phosphine (280 mg, 10 mol %) and potassium carbonate (1.28 g, 9.2 mmol) were added in a three-neck round bottom flask that was flushed with argon. Dry DMF (20 mL) was injected via a syringe and a greenish mixture formed. The mixture was heated at 85 °C for 2 h and a brown mixture afforded. The mixture was cooled to room temperature and poured into water (100 mL). The resulting mixture was extracted with ethyl acetate. The organic layer was washed thoroughly with water and brine to remove DMF and dried over magnesium sulfate. TLC showed the main product as a blue spot under short wavelength ultraviolet light. The red oil obtained after removal of solvent under reduced pressure was applied to a silica gel column and eluted with 50/50 hexane/methylene chloride. The major product was separated out as a yellow solid (1.38 g, 64%) after removal of solvent under reduced pressure. mp 87-90 °C;
1H-NMR (200 MHz, CDCl
3) δ 7.50-7.46 (m, 4H), 6.99-6.95 (dd, 2H, J=8.0 Hz, 2.0 Hz), 3.85 (s, 3H), 2.37 (s, 3H), 1.39 (s, 18H).
4-Acetoxy-3’-bromo-3,5-di-tert-butyl-4’-methoxybiphenyl (14)
Compound 13 (0.24 g, 0.68 mmol) was dissolved in dichloromethane (10 mL), and Br2 (0.33 g, 2 mmol) in dichloromethane (10mL) was added. The red-brown solution was refluxed under argon overnight. Saturated sodium bisulfite solution was added to quench excess bromine and the organic layer was washed with water and brine several times. The crude solid obtained after removal of solvent was recrystallized from pentane to give prism-shaped white crystals (0.17 g, 57%) with mp 141-143 °C; 1H-NMR (200 MHz, CDCl3) δ 7.73-7.72 (d, 1H, J=2.0 Hz), 7.48-7.43 (dd, s, 3H, J=2.0 Hz), 6.98-6.94 (d, 1H, J=8.0 Hz), 3.94 (s, 3H), 2.38 (s, 3H), 1.39 (s, 18H); Elemental analysis: calc’d for C23H29O3Br: C 63.74, H 6.70; found: C 63.73, H 6.69.
1,3-Bis(4-Acetoxy-3,5-di-tert-butyl-4’-methoxybiphenyl-3’-(E)-β-ethenyl)benzene (15)
Compound 14 (0.5 g, 1.15 mmol), palladium acetate (7.8 mg, 3 mol %) and tri-o-tolylphosphine (21 mg, 6 mol %) were placed in a 50-mL round bottom flask. The flask was evacuated for 10 min and refilled with argon. A solution of tributylamine (0.24 g, 1.3 mmol) and 1,3-divinylbenzene (0.075 g, 0.58 mmol) in DMF (20 mL) was then added via syringe. The resulting yellow mixture was heated to ca 110 °C under argon overnight and a dark green color was found. TLC showed disappearance of starting materials. The mixture was poured into water and extracted with ethyl acetate. The organic layer was washed thoroughly with water and brine several times and dried over magnesium sulfate. After evaporating the solvent, the residue was applied onto a silica gel column and eluted with 20/80 ethyl acetate/hexane. The material with highest Rf was isolated after removal of solvent to give a white crystalline solid (0.21 g, 44 %) that was suitable for use in the subsequent deprotection step to make 16. 1H-NMR (200 MHz, CDCl3) δ 7.76-6.94 (m, 18H), 3.95 (s, 6H), 2.39 (s, 6H), 1.41 (s, 36H); IR (KBr, cm-1): 1760, 960; UV-Vis (THF, λmax, nm): 214, 274, 3412.
1,3-Bis(3,5-Di-tert-butyl-4-hydroxy-4’-methoxybiphenyl-3’-(E)-ethenyl)benzene (16)
To a suspension of lithium aluminum hydride (0.1g, 2.6 mmol) in 10 mL of dry THF cooled in an ice bath, a solution of compound 15 (0.21 g, 0.25 mmol) in THF (5 mL) was added slowly via a cannula. A green suspension resulted and the reaction was allowed to stir for three hours at room temperature. The reaction was quenched by pouring into crushed ice and then acidified with hydrochloric acid. The resulting mixture was extracted with ethyl acetate. The organic layer was washed with water and brine, then dried over magnesium sulfate. An oily material was obtained after removal of solvent. Silica gel column chromatography with 20/80 ethyl acetate/hexane afforded a slightly greenish crystalline solid (0.1 g, 53 %) which sinters upon heating, mp 160 °C (dec). 1H-NMR (200 MHz, CDCl3) δ 7.73-6.95 (m, 18H), 5.25 (s, 2H), 3.94 (s, 6H), 1.51 (s, 36H). IR (neat, cm-1): 3600, 960. UV-Vis (benzene, λmax, nm): 284, 344. HRMS (EI, m/z): calc’d for C52H62O4: 750.4648; found: 750.4651.
Oxidation of compound 16
A small amount of compound 6 (ca. 5 mg) was dissolved in benzene (1 mL) under argon. Lead dioxide powder was added into this benzene solution and mixture was allowed to sit for a few minutes. A deep blue color was formed almost immediately. The final blue solution was separated from black lead dioxide by filtration. UV-Vis (benzene, λmax, nm): 319, 372, 614; ESR (9.8 GHz, benzene, gauss): a(H) = 1.36 (2H), 1.92 (2H).
2,6-Di-tert-butyl-4-vinylphenyl acetate (17)
To a suspension of CH
3PPh
3Br (15.25 g, 42.7 mmol) in THF (200 mL) cooled in an ice bath was added
n-butyllithium (27.5 mL of a 1.6 M in hexane, 44.0 mmol) via a syringe and a yellow mixture resulted. The mixture was warmed up to room temperature and stirred for additional 30 min. A solution of 3,5-di-
tert-butyl-4-hydroxybenzaldehyde (5.0 g, 21.4 mmol) in THF was added dropwise via a pressure-equalizing addition funnel. The resulted mixture was stirred for 2 h. The color of the mixture changed to blue after two hours. The mixture was recooled in an ice bath and acetyl chloride (3.35 g, 42.7 mmol) was injected into the reaction mixture with a syringe. The reaction was warmed up to room temperature. The resulting brown mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with water and dried over magnesium sulfate. Silica gel column chromatography (20:80 ethyl acetate/hexane) separated out the desired product as an orange crystalline solid (3.53 g, 60 %) with mp 59-61 °C (lit mp 67 °C [
22]);
1H-NMR (200 MHz, CDCl
3) δ 7.36 (s, 2H), 6.76-6.62 (dd, 1H, J=10.8 Hz, 17.3 Hz), 5.72-5.62 (dd, 1H, J=0.72 Hz, 17.3 Hz), 5.24-5.18 (dd, 1H, J=1.1 Hz, 10.8 Hz), 2.35 (s, 3H), 1.36 (s, 18H); IR (KBr, cm
-1): 1760, 998, 905.
1,3-Bis[2-(3,5-di-tert-butyl-4-acetyloxyphenyl)ethenyl]benzene (18)
Compound 17 (0.2 g, 0.73 mmol), 1,3-diiodobenzene (0.12 g, 0.37 mmol), palladium acetate (8.2 mg, 5 mol %) and tri-o-tolylphosphine (22.2 mg, 10 mol %) were placed into a three-neck round bottom flask, which was evacuated for 30 min and refilled with argon. Triethylamine (1 mL) and DMF (10 mL) were added via syringe. The yellow mixture was heated under argon at 100 °C. After overnight reaction, a dark colored mixture was produced. TLC showed that the starting materials were gone and that the main product spot fluoresced blue under short wavelength UV light. The reaction was quenched by pouring into water and the resulting mixture extracted with ethyl acetate. The organic layer was washed with dilute hydrochloric acid and then water several times to remove DMF. Careful separation by column chromatography (10:90 ethyl acetate/hexane) gave the desired product 18 as a yellowish solid (0.157 g, 70%) that was suitable for use in the subsequent deprotection step to make 19. 1H-NMR (200 MHz, CDCl3) δ 7.65-7.33 (m, 18H), 7.20-7.12 (d, 2H, J=16 Hz), 7.07-6.99 (d, 2H, J=16 Hz), 2.36 (s, 6H), 1.39 (s, 36H); IR (neat, cm-1): 2956, 2850, 1760, 960.
1,3-Bis[2-(3,5-di-tert-butyl-4-hydroxyphenyl)ethenyl]benzene (19)
To an ice cooled suspension of lithium aluminum hydride (0.055 g, 1.46 mmol) in dry THF (2 mL) was added dropwise a solution of compound 18 (0.157 g, 0.25 mmol) in dry THF (2 mL). The color of the suspension changed from grey to yellowish and it was allowed to stir at room temperature for an additional 4 h. The mixture was poured into ice, acidified with conc HCl and extracted with ethyl acetate. The organic layer was washed with water and dried over magnesium sulfate. A yellow oil was obtained after removal of the solvent. Silica gel column chromatography gave a fraction that contained mainly the desired product with some impurities. Slow evaporation of the solvent allowed growth of yellow crystals (0.1 g, 74 %, mp 105 °C[d]) that showed only one spot by TLC. 1H‑NMR (200 MHz, CDCl3) δ 7.63 (m, 1H), 7.37 (m, 7H), 7.18-7.10 (d, 2H, J=16), 7.00-6.92 (d, 2H, J=16), 5.30 (s, 2H), 1.49 (s, 36H); IR (neat, cm-1): 3638, 2956, 2871, 957; UV-Vis (benzene, λmax, nm): 328 nm; Elemental analysis: calc’d for C38H50O2: C 84.76, H 9.29; found: C 85.20, H 9.69.
Oxidation of 19
A small amount of compound 19 (ca. 5 mg) was dissolved in dry benzene (1 mL) under argon. To this solution was added lead dioxide powder. Color changed from colorless to dark yellow. Lead dioxide was filtered off and the filtrate was subjected ESR spectroscopy. IR showed disappearance of the sharp OH stretch at 3640 cm-1 that is observed in compound 19. UV-Vis (benzene, λmax, nm): 309, 457, 488; ESR (9.751 GHz, benzene, gauss): a(H) = 6.37, 2.93, 1.69, 1.59, 1.48, 1.47, 1.40, 0.53.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}