Measurement of Nanomolar Dissociation Constants by Titration Calorimetry and Thermal Shift Assay – Radicicol Binding to Hsp90 and Ethoxzolamide Binding to CAII

Abstract
:1. Introduction
2. Results and Discussion
2.1. Derivation of the 2-stage thermal shift assay binding model
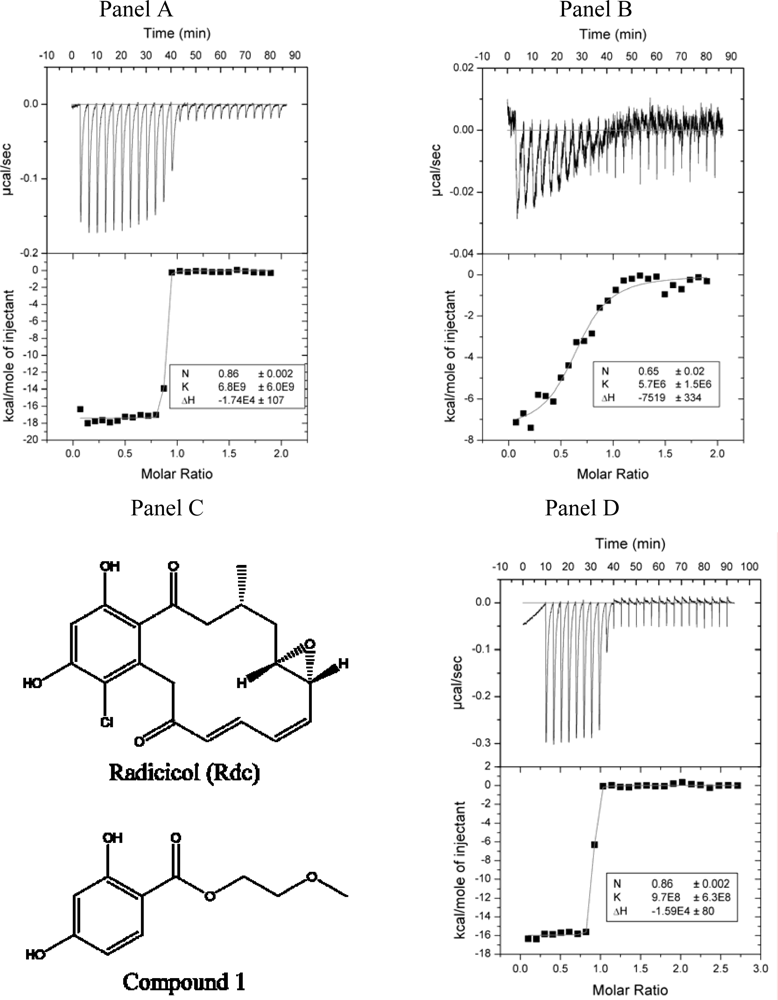
2.2. Radicicol binding to Hsp90 and ethoxzolamide binding to hCAII by titration calorimetry
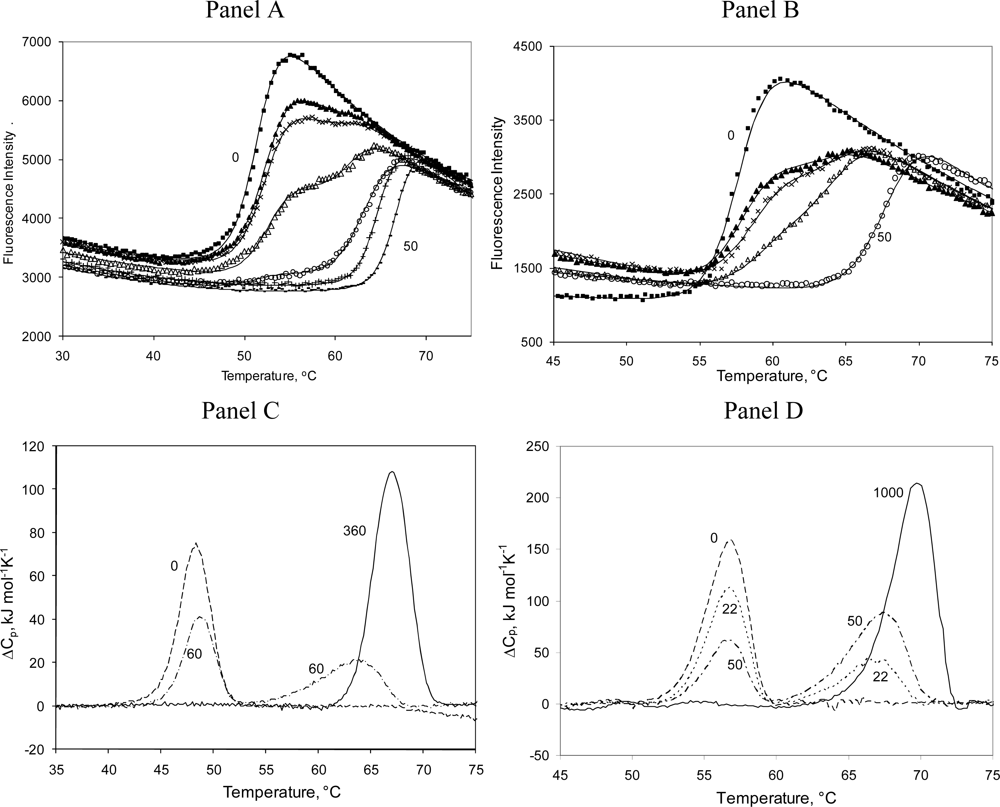
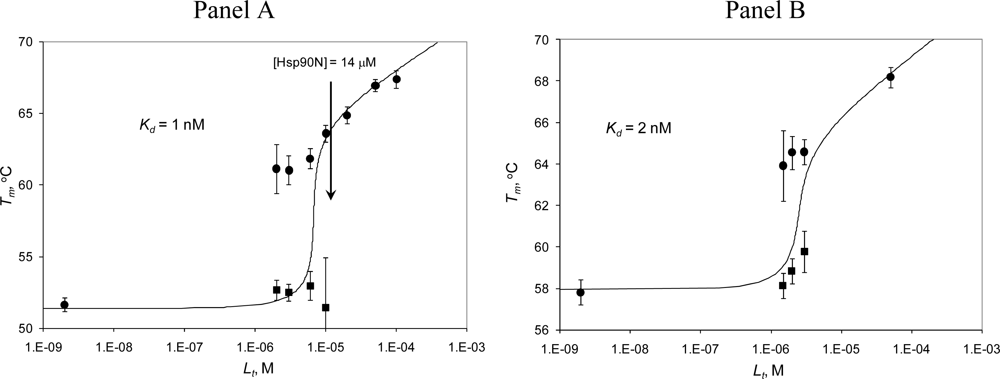
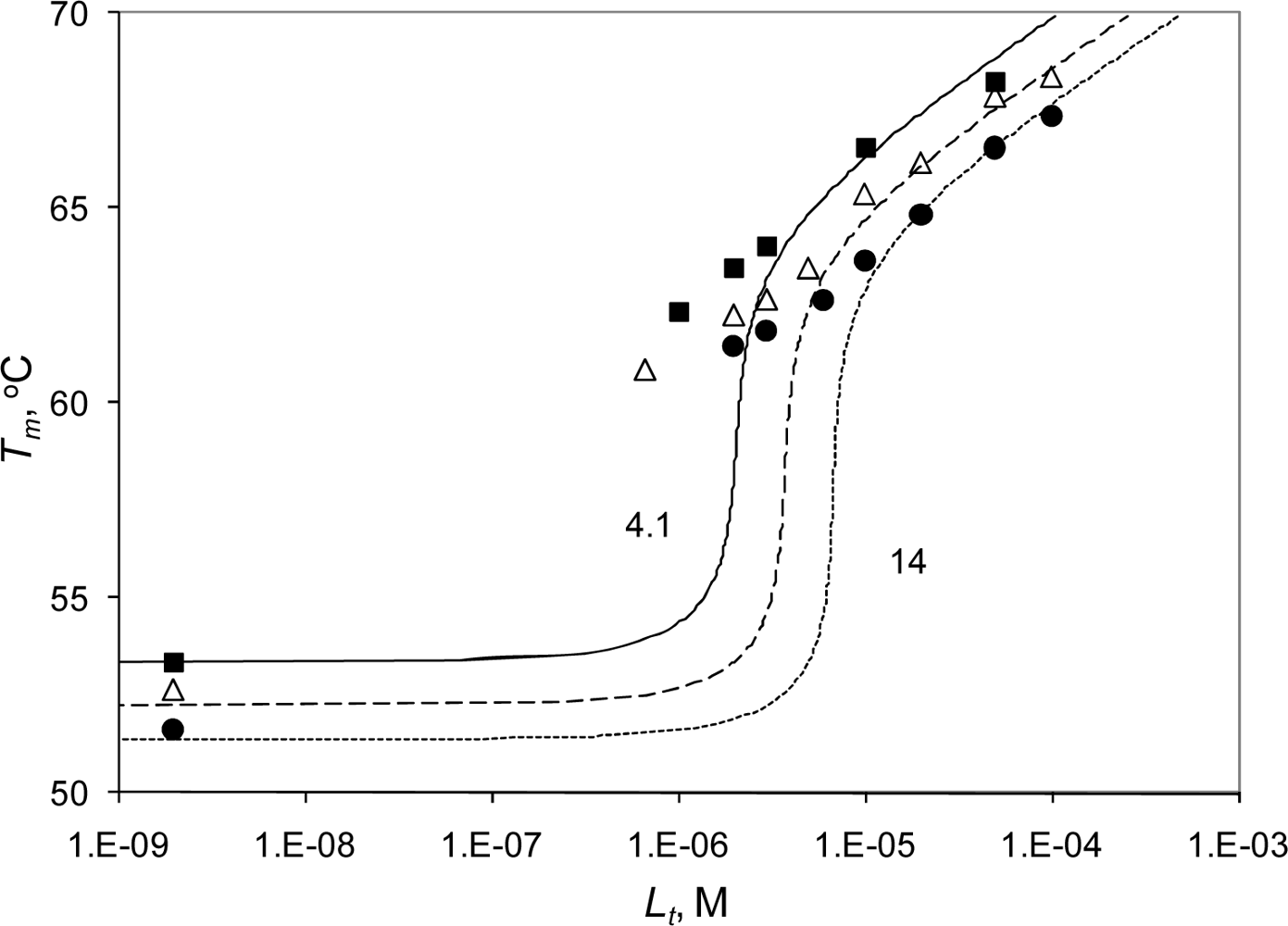
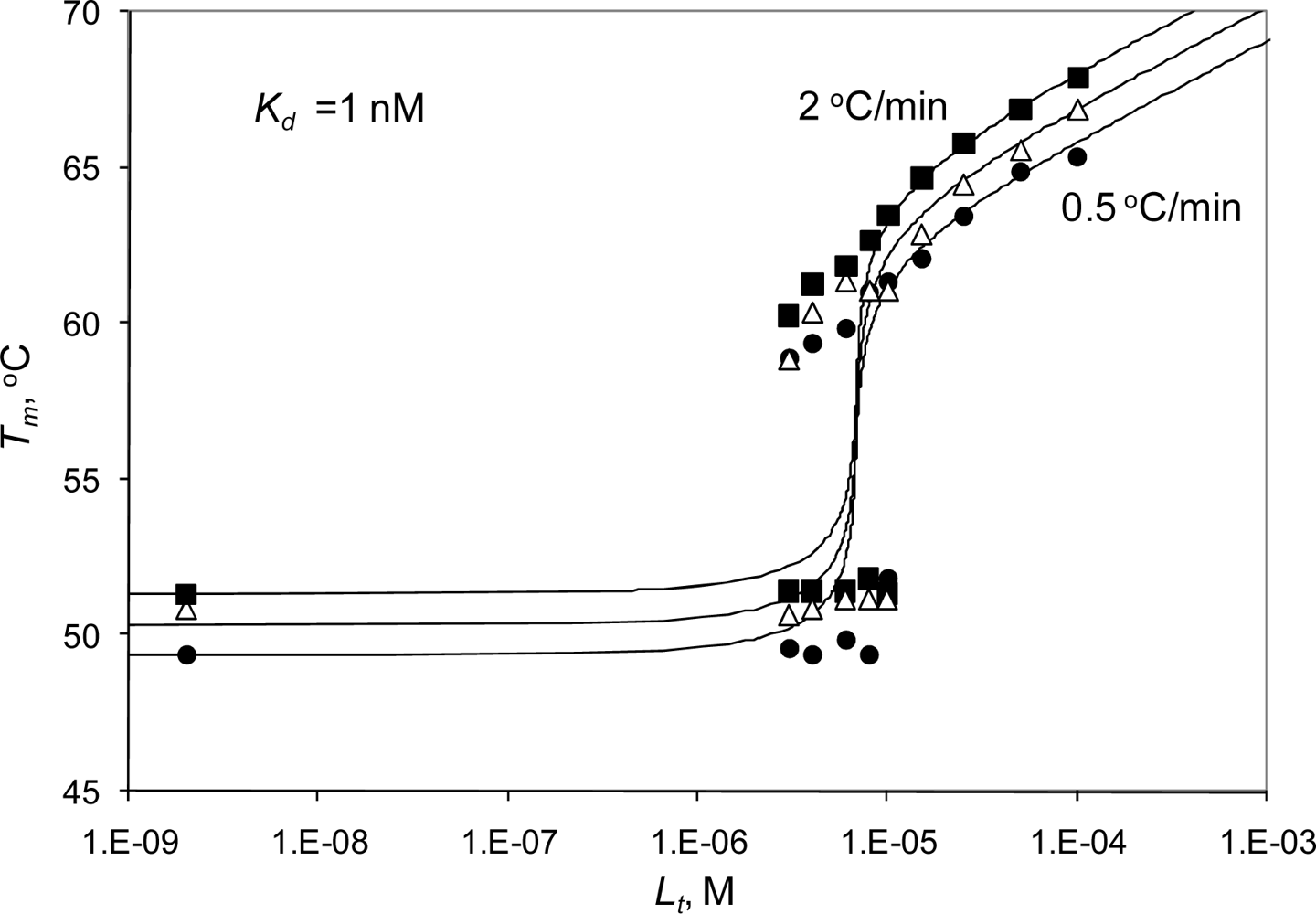
2.3. Radicicol binding to Hsp90 and ethoxzolamide binding to hCAII by TSA and DSC
2.4. Discussion
3. Experimental Section
3.1. Materials
3.2. Hsp90 constructs
3.3. Protein expression and purification
3.4. Production of recombinant human carbonic anhydrase II
3.5. Mass spectrometry experiments
3.6. Isothermal titration calorimetry
3.7. Protein denaturation experiments
3.8. Differential Scanning Calorimetry (DSC) experiments
4. Conclusions
Acknowledgments
References and Notes
- Sigurskjold, BW. Exact analysis of competition ligand binding by displacement isothermal titration calorimetry. Anal Biochem 2000, 277(2), 260–266. [Google Scholar]
- Velazquez-Campoy, A; Freire, E. Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat Protoc 2006, 1(1), 186–191. [Google Scholar]
- Lo, MC; Aulabaugh, A; Jin, G; Cowling, R; Bard, J; Malamas, M; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 2004, 332(1), 153–159. [Google Scholar]
- Niesen, FH; Berglund, H; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2007, 2(9), 2212–2221. [Google Scholar]
- Pantoliano, MW; Petrella, EC; Kwasnoski, JD; Lobanov, VS; Myslik, J; Graf, E; Carver, T; Asel, E; Springer, BA; Lane, P; Salemme, FR. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen 2001, 6(6), 429–440. [Google Scholar]
- Todd, MJ; Salemme, FR. Direct binding assays for pharma screening. Genetic Eng News 2003, 23(3), 28–29. [Google Scholar]
- Cimmperman, P; Baranauskiene, L; Jachimoviciute, S; Jachno, J; Torresan, J; Michailoviene, V; Matuliene, J; Sereikaite, J; Bumelis, V; Matulis, D. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys J 2008, 95(7), 3222–3231. [Google Scholar]
- Mezzasalma, TM; Kranz, JK; Chan, W; Struble, GT; Schalk-Hihi, C; Deckman, IC; Springer, BA; Todd, MJ. Enhancing recombinant protein quality and yield by protein stability profiling. J Biomol Screen 2007, 12(3), 418–428. [Google Scholar]
- Ericsson, UB; Hallberg, BM; Detitta, GT; Dekker, N; Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem 2006, 357(2), 289–298. [Google Scholar]
- Brandts, JF; Lin, LN. Study of strong to ultratight protein interactions using differential scanning calorimetry. Biochemistry 1990, 29(29), 6927–6940. [Google Scholar]
- Shrake, A; Ross, PD. Biphasic denaturation of human albumin due to ligand redistribution during unfolding. J Biol Chem 1988, 263(30), 15392–15399. [Google Scholar]
- Anderson, SR; Weber, G. Fluorescence polarization of the complexes of 1-anilino-8-naphthalenesulfonate with bovine serum albumin. Evidence for preferential orientation of the ligand. Biochemistry 1969, 8(1), 371–377. [Google Scholar]
- Weber, G; Laurence, DJ. Fluorescent indicators of adsorption in aqueous solution and on the solid phase. Process Biochem 1954, 56, xxxi. [Google Scholar]
- Slavik, J; Horak, J; Rihova, L; Kotyk, A. Anilinonaphthalene sulfonate fluorescence and amino acid transport in yeast. J Membr Biol 1982, 64(3), 175–179. [Google Scholar]
- Matulis, D; Baumann, CG; Bloomfield, VA; Lovrien, RE. 1-Anilino-8-naphthalene sulfonate as a protein conformational tightening agent. Biopolymers 1999, 49(6), 451–458. [Google Scholar]
- Matulis, D; Lovrien, R. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys J 1998, 74(1), 422–429. [Google Scholar]
- Matulis, D; Kranz, JK; Salemme, FR; Todd, MJ. Thermodynamic stability of carbonic anhydrase: Measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 2005, 44(13), 5258–5266. [Google Scholar]
- Maroney, AC; Marugan, JJ; Mezzasalma, TM; Barnakov, AN; Garrabrant, TA; Weaner, LE; Jones, WJ; Barnakova, LA; Koblish, HK; Todd, MJ; Masucci, JA; Deckman, IC; Galemmo, RA, Jr; Johnson, DL. Dihydroquinone ansamycins: Toward resolving the conflict between low in vitro affinity and high cellular potency of geldanamycin derivatives. Biochemistry 2006, 45(17), 5678–5685. [Google Scholar]
- Shrake, A; Ross, PD. Origins and consequences of ligand-induced multiphasic thermal protein denaturation. Biopolymers 1992, 32(8), 925–940. [Google Scholar]
- Shrake, A; Ross, PD. Ligand-induced biphasic protein denaturation. J Biol Chem 1990, 265(9), 5055–5059. [Google Scholar]
- Wandinger, SK; Richter, K; Buchner, J. The Hsp90 chaperone machinery. J Biol Chem 2008, 283(27), 18473–18477. [Google Scholar]
- Richter, K; Meinlschmidt, B; Buchner, J. Hsp90: From dispensable heat shock protein to global player. In Protein Folding Handbook; Buchner, J, Kiefhaber, T, Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2005; pp. 768–829. [Google Scholar]
- Pearl, LH; Prodromou, C; Workman, P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J 2008, 410(3), 439–453. [Google Scholar]
- Blagg, BS; Kerr, TD. Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med Res Rev 2006, 26(3), 310–338. [Google Scholar]
- Drysdale, MJ; Brough, PA; Massey, A; Jensen, MR; Schoepfer, J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel 2006, 9(4), 483–495. [Google Scholar]
- Whitesell, L; Lindquist, SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005, 5(10), 761–772. [Google Scholar]
- Solit, DB; Chiosis, G. Development and application of Hsp90 inhibitors. Drug Discov Today 2008, 13(1–2), 38–43. [Google Scholar]
- Ali, MM; Roe, SM; Vaughan, CK; Meyer, P; Panaretou, B; Piper, PW; Prodromou, C; Pearl, LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440(7087), 1013–1017. [Google Scholar]
- Delmotte, P; Delmotte-Plaque, J. A new antifungal substance of fungal origin. Nature 1953, 171(4347), 344. [Google Scholar]
- Schulte, TW; Akinaga, S; Soga, S; Sullivan, W; Stensgard, B; Toft, D; Neckers, LM. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998, 3(2), 100–108. [Google Scholar]
- Roe, SM; Prodromou, C; O'Brien, R; Ladbury, JE; Piper, PW; Pearl, LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 1999, 42(2), 260–266. [Google Scholar]
- Sharp, SY; Boxall, K; Rowlands, M; Prodromou, C; Roe, SM; Maloney, A; Powers, M; Clarke, PA; Box, G; Sanderson, S; Patterson, L; Matthews, TP; Cheung, KM; Ball, K; Hayes, A; Raynaud, F; Marais, R; Pearl, L; Eccles, S; Aherne, W; McDonald, E; Workman, P. In vitro biological characterization of a novel, synthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors. Cancer Res 2007, 67(5), 2206–2216. [Google Scholar]
- Supuran, CT. Carbonic anhydrases - an overview. Curr Pharm Des 2008, 14(7), 603–614. [Google Scholar]
- Supuran, CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008, 7(2), 168–181. [Google Scholar]
- Thoms, S. Hydrogen bonds and the catalytic mechanism of human carbonic anhydrase II. J Theor Biol 2002, 215(4), 399–404. [Google Scholar]
- Di Fiore, A; Pedone, C; Antel, J; Waldeck, H; Witte, A; Wurl, M; Scozzafava, A; Supuran, CT; De Simone, G. Carbonic anhydrase inhibitors: the X-ray crystal structure of ethoxzolamide complexed to human isoform II reveals the importance of thr200 and gln92 for obtaining tight-binding inhibitors. Bioorg Med Chem Lett 2008, 18(8), 2669–2674. [Google Scholar]
- Garnier, C; Protasevich, I; Gilli, R; Tsvetkov, P; Lobachov, V; Peyrot, V; Briand, C; Makarov, A. The two-state process of the heat shock protein 90 thermal denaturation: Effect of calcium and magnesium. Biochem Biophys Res Commun 1998, 249(1), 197–201. [Google Scholar]
- Agatsuma, T; Ogawa, H; Akasaka, K; Asai, A; Yamashita, Y; Mizukami, T; Akinaga, S; Saitoh, Y. Halohydrin and oxime derivatives of radicicol: synthesis and antitumor activities. Bioorg Med Chem 2002, 10(11), 3445–3454. [Google Scholar]
- Robertson, AD; Murphy, KP. Protein structure and the energetics of protein stability. Chem Rev 1997, 97(5), 1251–1268. [Google Scholar]
- Rosgen, J; Hinz, HJ. Phase diagrams: a graphical representation of linkage relations. J Mol Biol 2003, 328(1), 255–271. [Google Scholar]
- Casini, A; Antel, J; Abbate, F; Scozzafava, A; David, S; Waldeck, H; Schafer, S; Supuran, CT. Carbonic anhydrase inhibitors: SAR and X-ray crystallographic study for the interaction of sugar sulfamates/sulfamides with isozymes I, II and IV. Bioorg Med Chem Lett 2003, 13(5), 841–845. [Google Scholar]
- Baranauskiene, L; Matuliene, J; Matulis, D. Determination of the thermodynamics of carbonic anhydrase acid-unfolding by titration calorimetry. J Biochem Biophys Methods 2008, 70(6), 1043–1047. [Google Scholar]
- Sigg, HP; Loeffler, W. Production of radicicol. US Pat 3428526 1969. [Google Scholar]
- Richter, K; Muschler, P; Hainzl, O; Buchner, J. Coordinated ATP hydrolysis by the Hsp90 dimer. J Biol Chem 2001, 276(36), 33689–33696. [Google Scholar]
Abbreviations:
| ANS: | 1,8-anilinonaphthalene sulfonate; |
| DMSO: | dimethyl sulfoxide; |
| DSC: | differential scanning calorimetry; |
| DTT: | 1,4-dithiothreitol; |
| bCAII: | bovine carbonic anhydrase II; |
| hCAII: | recombinant human carbonic anhydrase II; |
| Hsp90αN: | N-terminal domain of recombinant human alpha Hsp90; |
| Hsp90: | heat shock protein 90; |
| ITC: | isothermal titration calorimetry; |
| Rdc: | radicicol; |
| Tris: | tris(hydroxymethyl)-aminomethane; |
| TSA: | thermal shift assay |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Ligand], μM | Tfree, ºC | Tbound, ºC | ΔuHfree, kJ/mol | ΔuHbound, kJ/mol | n |
|---|---|---|---|---|---|
| Uncertainties- | ±1.2 | ±1.2 | ±140(TSA)
±30 (DSC) | ±140 (TSA)
±30 (DSC) | ±0.15 |
| Hsp90αN (14 μM) with radicicol (TSA) | |||||
| 0 | 51.6 | – | 670 | – | 0 |
| 2 | 52.7 | 61.1 | 650 | 610 | 0.15 |
| 3 | 52.5 | 61.0 | 610 | 610 | 0.22 |
| 6 | 53.0 | 61.8 | 600 | 600 | 0.44 |
| 10 | 51.4 | 63.6 | 610 | 610 | 0.74 |
| 20 | – | 64.9 | – | 870 | 1 |
| 50 | – | 66.9 | – | 900 | 1 |
| Hsp90αN (120 μM) with radicicol (DSC) | |||||
| 0 | 48.2 | – | 290 | – | 0 |
| 60 | 48.6 | 63.4 | 302 | 283 | 0.5 |
| 360 | – | 66.9 | – | 477 | 1 |
| hCAII (5 μM) with ethoxzolamide (TSA) | |||||
| 0 | 57.8 | – | 690 | – | 0 |
| 1.5 | 58.1 | 63.9 | 720 | 750 | 0.3 |
| 2 | 58.8 | 64.6 | 660 | 720 | 0.4 |
| 3 | 59.8 | 64.6 | 610 | 740 | 0.6 |
| 50 | – | 68.2 | – | 780 | 1 |
| hCAII (100 μM) with ethoxzolamide (DSC) | |||||
| 0 | 56.5 | – | 570 | – | 0 |
| 22 | 56.5 | 66.5 | 490 | 890 | 0.2 |
| 50 | 56.4 | 66.9 | 420 | 860 | 0.5 |
| 1000 | – | 69.4 | – | 790 | 1 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zubrienė, A.; Matulienė, J.; Baranauskienė, L.; Jachno, J.; Torresan, J.; Michailovienė, V.; Cimmperman, P.; Matulis, D. Measurement of Nanomolar Dissociation Constants by Titration Calorimetry and Thermal Shift Assay – Radicicol Binding to Hsp90 and Ethoxzolamide Binding to CAII. Int. J. Mol. Sci. 2009, 10, 2662-2680. https://doi.org/10.3390/ijms10062662
Zubrienė A, Matulienė J, Baranauskienė L, Jachno J, Torresan J, Michailovienė V, Cimmperman P, Matulis D. Measurement of Nanomolar Dissociation Constants by Titration Calorimetry and Thermal Shift Assay – Radicicol Binding to Hsp90 and Ethoxzolamide Binding to CAII. International Journal of Molecular Sciences. 2009; 10(6):2662-2680. https://doi.org/10.3390/ijms10062662
Chicago/Turabian StyleZubrienė, Asta, Jurgita Matulienė, Lina Baranauskienė, Jelena Jachno, Jolanta Torresan, Vilma Michailovienė, Piotras Cimmperman, and Daumantas Matulis. 2009. "Measurement of Nanomolar Dissociation Constants by Titration Calorimetry and Thermal Shift Assay – Radicicol Binding to Hsp90 and Ethoxzolamide Binding to CAII" International Journal of Molecular Sciences 10, no. 6: 2662-2680. https://doi.org/10.3390/ijms10062662
APA StyleZubrienė, A., Matulienė, J., Baranauskienė, L., Jachno, J., Torresan, J., Michailovienė, V., Cimmperman, P., & Matulis, D. (2009). Measurement of Nanomolar Dissociation Constants by Titration Calorimetry and Thermal Shift Assay – Radicicol Binding to Hsp90 and Ethoxzolamide Binding to CAII. International Journal of Molecular Sciences, 10(6), 2662-2680. https://doi.org/10.3390/ijms10062662