1. Introduction
Parkinson’s disease (PD) is clearly defined as a neurodegenerative disorder that involves multifactorial mechanisms, such as mitochondrial dysfunction, neuronal apoptosis, neuroinflammation and microglial activation, which leads to the progressive loss of dopamine-producing cells in the nigro-striatal system [
1,
2]. Unfortunately, the conventional therapy for PD focuses primarily on the consequences of this neuronal loss: the stimulation of degenerating dopamine cells [
3]. Therefore, novel pharmacotherapeutic strategies are being developed that focus on cellular targets to prevent or delay dopamine-induced cell death at much earlier stages of neurodegeneration. These approaches may reveal novel strategies for the treatment of PD.
Mitochondria can be considered one of these novel drug targets. There are several arguments supporting the view that genetic defects in mitochondria play a prevalent role in the pathogenesis of PD [
4–
6]. Defects in complex I are considered a central and early cause of PD [
4] because a deficiency in complex I activity initiates further respiratory chain dysfunctions that involve mitochondria-dependent apoptotic cascades and result in cell death [
7].
Therefore, the protection of mitochondria, even their repair mechanisms at the level of complex I, may be a key strategy in limiting mitochondrial damage and ensuring cellular integrity [
8]. At present, drugs that provide such effect have not been found.
A more complicated problem is the determination of ways to control the development of pathological changes in protein conformations that are associated with PD. Mutations in the genes that encode a series of proteins, particularly α-synuclein (SNCA), parkin, DJ-1, PINK1, LRRK2 and HTR2A, lead to the formation of misfolded protein conformations that are harmful to cells [
1,
2]. The aggregation of α-synuclein in Lewy bodies plays a crucial role in cell death because such aggregates activate pro-inflammatory molecules and switch on the neuroinflammatory and apoptotic events that lead to cell death. Recent evidence suggests that misfolded proteins and their aggregates are closely related to the activities of endogenous or molecular chaperones, which are intracellular molecules that are capable of unfolding proteins [
9]. Among these chaperones, the heat shock protein (HSP) family is the most commonly studied group.
Decreasing levels of HSP and α-synuclein aggregation in Lewy bodies coincide with the progression of PD [
10]. Experimental data show that HSP70 may inhibit α-synuclein fibril formation [
11] and possess anti-apoptotic activity [
10].
Designing chemical or pharmacological chaperones (pharmacoperons) that can unfold misfolded proteins in a similar manner to endogenous molecules is an attractive approach for future PD therapies [
12]. Numerous molecules have been shown to possess the properties of chemical chaperones, such as glycerol, diltiazem, vinblastine, trimethylamine
N-oxide, rifampicin, flavonoids and glycine betaine [
13–
17].
A very important consequence of a build-up and aggregation of misfolded proteins is the impairment of the ubiquitin-proteasome degradation system (UPS) [
10]. Normally, proteins that are conjugated to ubiquitin are targeted to the UPS where they undergo proteolytic degradation [
18]. The ubiquitin/proteasome pathway is the major proteolytic quality control system in cells, and ubiquitin-enriched Lewy bodies in dopaminergic neurons are a hallmark of PD [
19]. A high level of ubiquitin and ubiquitinated proteins (
i.e., ubiquitin that is covalently attached to target proteins) in Lewy bodies indicates that protein degradation by proteasomes is impaired [
20]. Therefore, ubiquitination pathways may play a central role in the pathogenesis of PD [
18], and the regulation of the level of ubiquitin may serve as another drug target.
Others studies emphasize glial cells as a target for the treatment of PD. Microglial activation plays an important role in the early stages (before dopaminergic cell death) of PD pathogenesis [
21]. This activation triggers the onset of a molecular cascade that leads to the progressive degeneration of cells [
22]. Therefore, the blockade of microglial activation might be a target for therapeutic intervention in PD [
23]. Moreover, a new concept of glia as a stem cell element in the brain has been recently proposed [
24]. Furthermore, astrocytes are involved in the formation of synapses and in the modulation of synaptic functions via a bidirectional communication with neurons [
25]. Brain damage caused by stroke, degeneration or demyelination may elicit reactive astrogliosis, which has been suggested to impede neuronal survival [
23]. Conversely, activated astrocytes may promote neurogenesis by producing energy-producing compounds and neurotrophins [
26,
27]. Neurotrophins, such as glial cell line-derived neurotrophic factor (GDNF) [
28] and mesencephalic astrocyte-derived neurotrophic factor (MANF) [
29], have been proposed to promote the survival and differentiation of dopamine neurons. Nevertheless, their clinical use is limited by their large molecular size and antigenicity.
In this context, the idea of adult neurogenesis as a potential tool for the restoration of lost dopamine neurons might open new vistas for the development of novel therapeutics in PD [
30]. Although neural or fetal stem cell transplantation has been used previously to replace damaged neurons [
31], the stimulation of endogenous progenitor cells is an exciting alternative [
32]. In PD brain structures, a decrease in progenitor cell proliferation has been demonstrated [
33]. In a 6-OHDA-lesioned PD model in rats, an increase in progenitor cell proliferation in nigro-striatal structures has been noted [
32]. The search for drugs that are capable of regulating progenitor cell proliferation in the treatment of PD is a very new and critical issue. Guanosine was recently shown to induce the proliferation of neural progenitor stem cells in the adult SN and subventricular zone in a proteasome inhibitor-induced PD model in rats [
34].
Small molecules that are capable of altering the structure of proteins may be optimal pharmacophores (e.g., positively charged moieties) and serve as privileged molecules that can bind easily to the negatively charged amino acids of cellular proteins and target protein-protein interactions. Therefore, these small molecules may affect protein conformations and act as multivalent regulators of intracellular processes.
Small molecules have been suggested to function as multi-protein co-activators of gene transcription [
35]. Therefore, small molecules may demonstrate a beneficial protective effect in PD by slowing or halting the progression of the disease during its early stages.
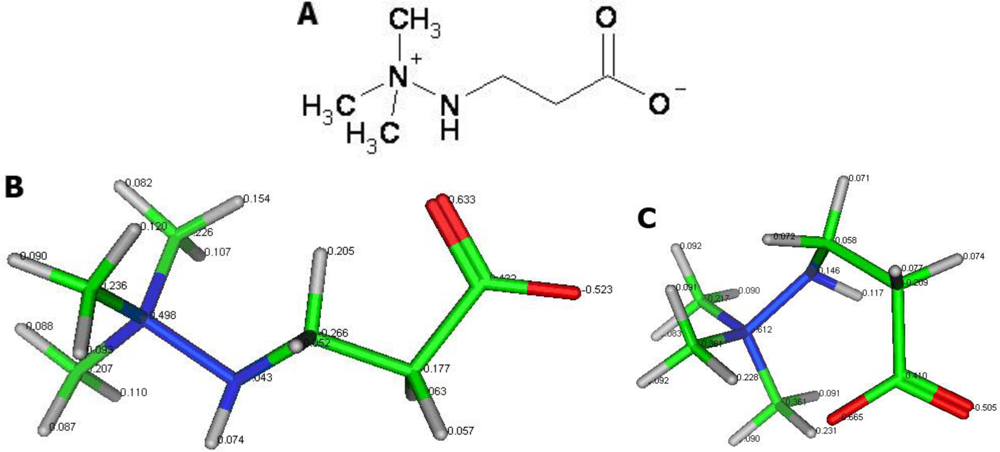
We propose that mildronate (
Figure 1), a representative of the aza-butyrobetaine class of compounds, partially meets the requirements for small molecules. Mildronate possesses a quaternary nitrogen and a negatively charged oxygen. These charged moieties might change the conformation of mildronate from a linear to a cyclic one via intramolecular electrostatic interactions. Cyclic mildronate is a neutral molecule, which allows it to penetrate the brain. Recently, an HPLC method was used to detect mildronate at a concentration of 102 ± 22 nmol/g in brain tissue after a 21 day intraperitoneal administration of 100 mg/kg [
36]. In an azidothymidine neurotoxicity model in rats, mildronate suppressed the neuroinflammatory and apoptotic processes in brain tissue by protecting the abnormal expression of cytochrome oxidase c, caspase-3, iNOS, cellular apoptosis susceptibility (CAS) protein and glial fibrillary acidic protein (GFAP) [
37]. In addition, data from our previous studies have shown that mildronate plays a protective role against mitochondrial complex I damage caused by mitochondria-compromising substances, such as rotenone and the anti-HIV drug azidothymidine [
38].
The present study investigated the effects of mildronate in a 6-OHDA rat model of PD by assessing cellular biomarkers that are involved in signaling cascades and that are crucial for neural and glial integration: the neuronal marker TH (tyrosine hydroxylase); ubiquitin (a regulatory peptide involved in the ubiquitin-proteasome degradation system); Notch-3 (a marker of progenitor cells); IBA-1 (ionised calcium-binding adaptor molecule 1, a marker of microglia); glial fibrillary acidic protein, GFAP (a marker of astrocytes); inducible nitric oxide synthase, iNOS (a marker of inflammation).
3. Discussion
Recent new data demonstrate that macroglial and microglial cells contribute considerably to homeostasis in the brain [
25]. Therefore, novel therapeutic strategies for the treatment of PD can be rationally focused on both neuronal and glial processes. Mitochondria, which play a critical role in the aetiology of PD [
39], can be considered one of the most essential targets that are common to both neurons and glial cells. The oxidation rates of various substrates and the acceptor control ratios do not differ appreciably between neuronal and glial cell mitochondria [
40]. In this context, mitochondria are vulnerable to the effects of 6-OHDA-induced cell death [
41]. Moreover, 6-OHDA has been proposed to be a putative endogenous neurotoxic factor in the pathogenesis of PD because it can be generated from dopamine via non-enzymatic hydroxylation processes in the presence of Fe
2+ and H
2O
2 [
42].
We suggest that a small molecule, mildronate, might function as a suitable “interface molecule” between neuronal and glial systems and correct the imbalances that are present in the rat model of PD. This assumption was based on our previous findings that demonstrated the protective effects of mildronate at the level of the mitochondrial respiratory chain complex I [
38] and the ability of mildronate to exert anti-inflammatory and anti-apoptotic effects in a rat model of neurotoxicity generated with the mitochondria-compromising drug azidothymidine [
37]. In the present study, we assessed numerous markers of impaired neuronal and glial cell functions in the nigro-striatal system.
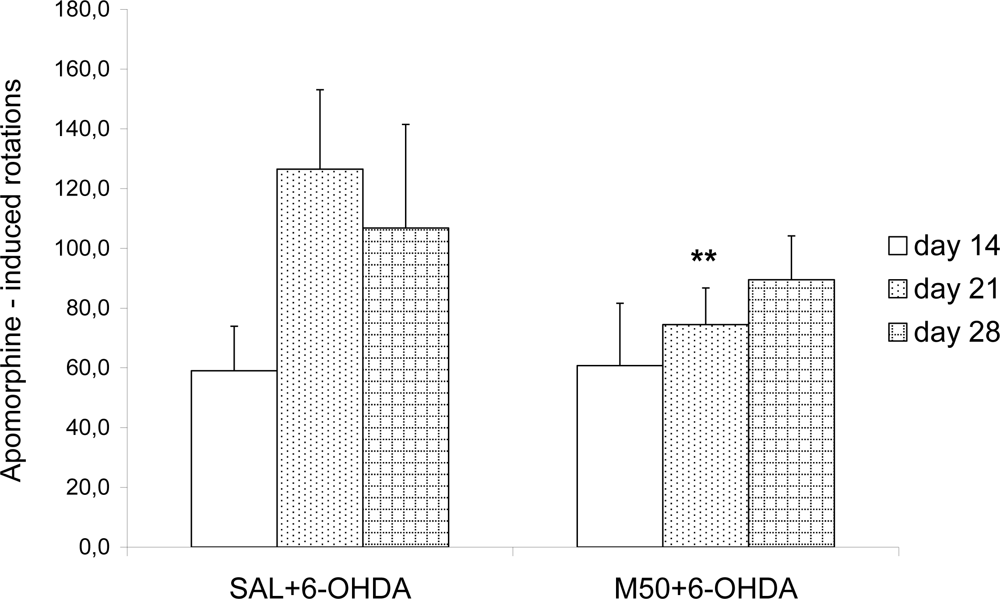
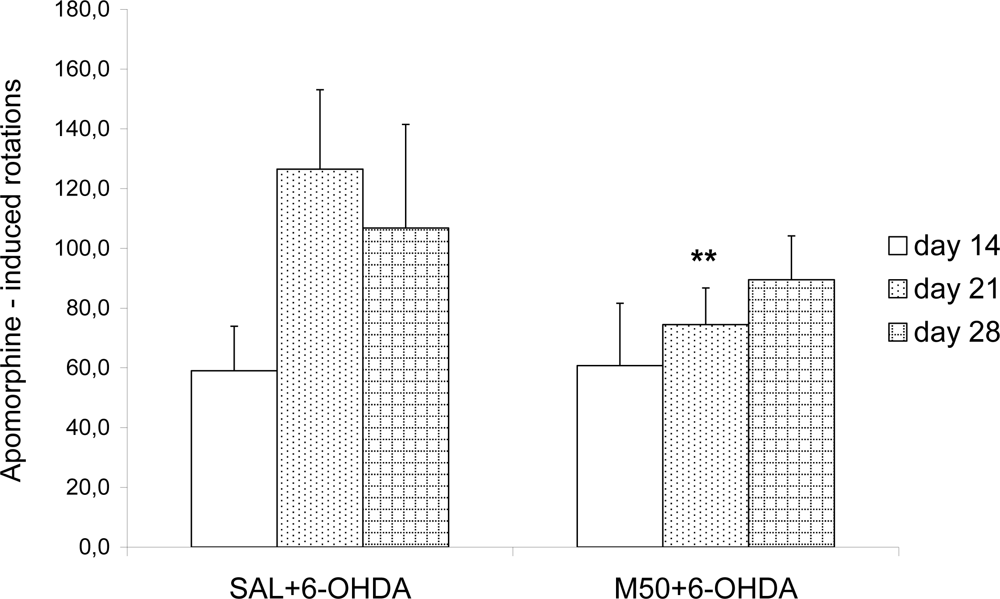
In our study the rotations behavior test showed that both doses of mildronate decreased the apomorphine rotations in 6-OHDA-treated rats, particularly on day 21, which indicated that mildronate attenuated behavioral disturbances in a PD model.
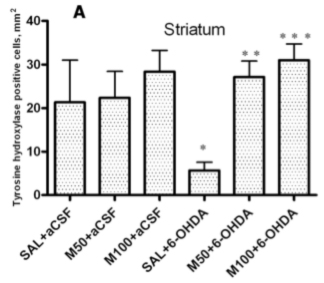
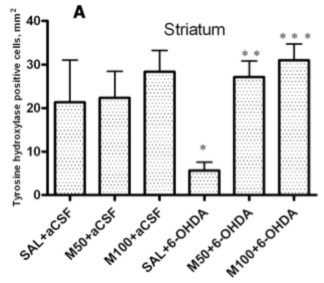
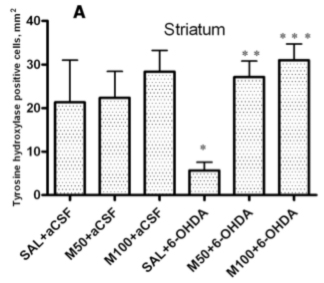
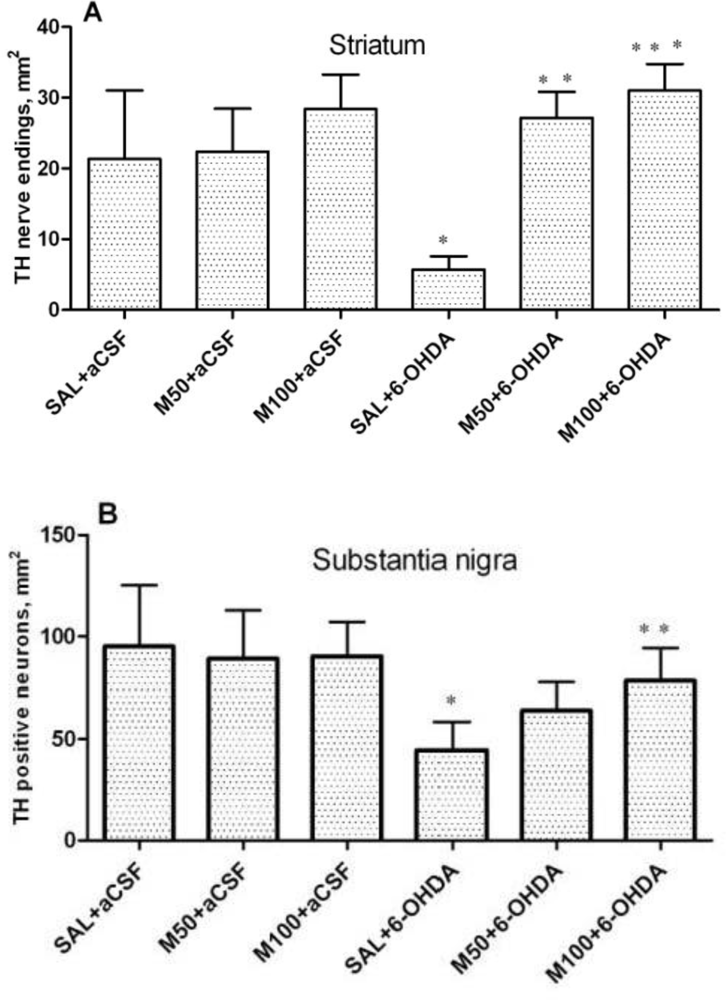
An immunohistochemical examination of brain tissue ex vivo demonstrated that 6-OHDA-induced lesions were associated with a massive loss of neuronal dopamine, a dramatic (five-fold) decrease in the number of TH-positive nerve endings in the lesioned striatum and an approximately 2.5-fold decrease in the number of TH-positive neurons in the SN in comparison to the control group (saline + aCSF). The administration of mildronate at a dose of 50 mg/kg provided complete protection against the 6-OHDA-induced degeneration of TH-positive nerve endings in the lesioned striatum. In addition, mildronate at 100 mg/kg protected against the loss of TH-positive dopaminergic neurons in the SN.
Our results are consistent with previous findings demonstrating that 6-OHDA decreased both the number of TH-positive nerve endings in the striatum and the number of TH dopaminergic neurons in the SN [
23]. Therefore, our data demonstrate the neuroprotective properties of mildronate against a 6-OHDA-induced loss of TH expression. These findings raise the following question: what are the crucial mechanisms responsible for this protective action of mildronate?
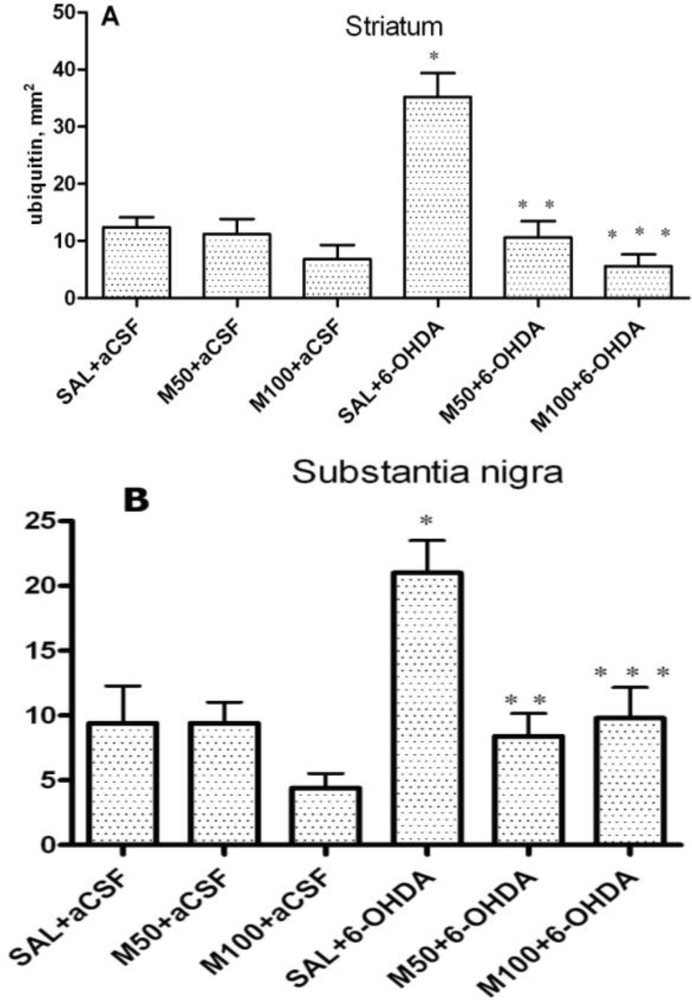
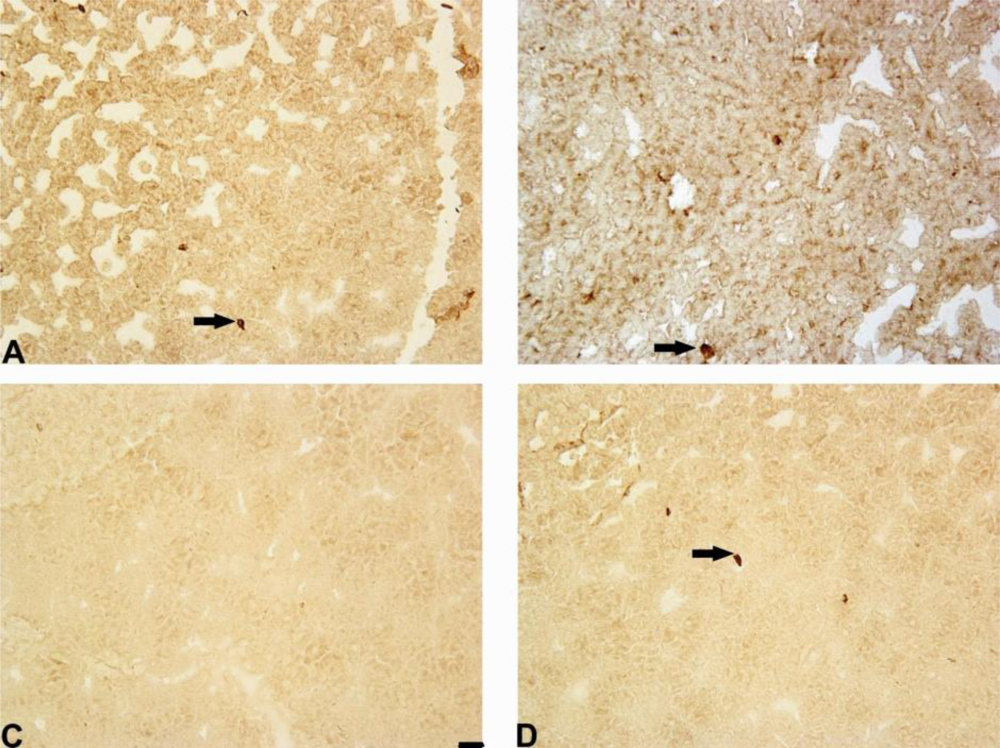
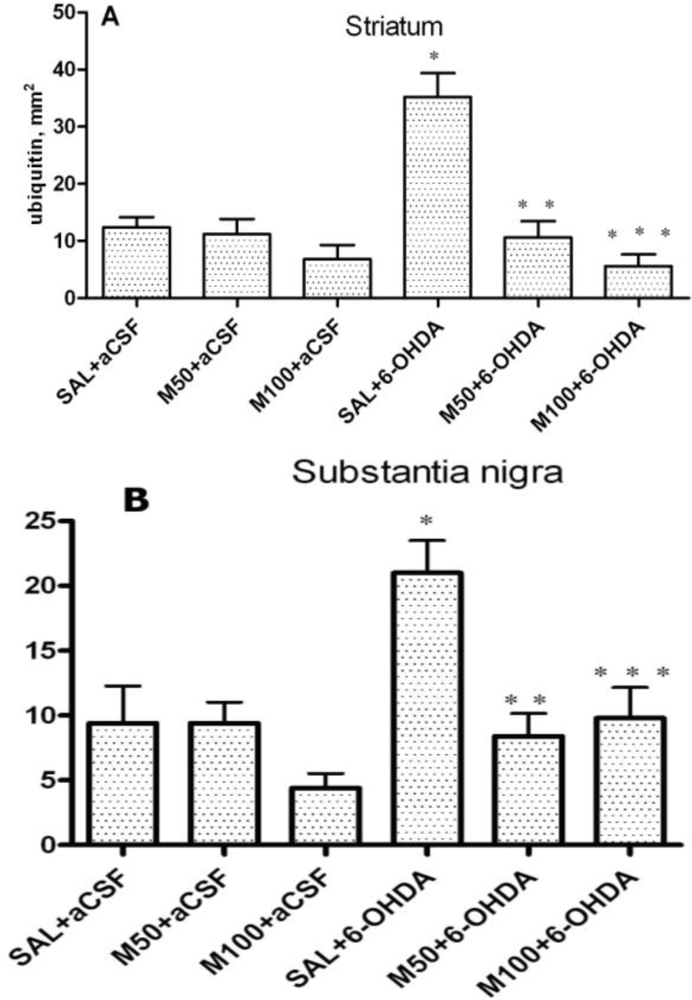
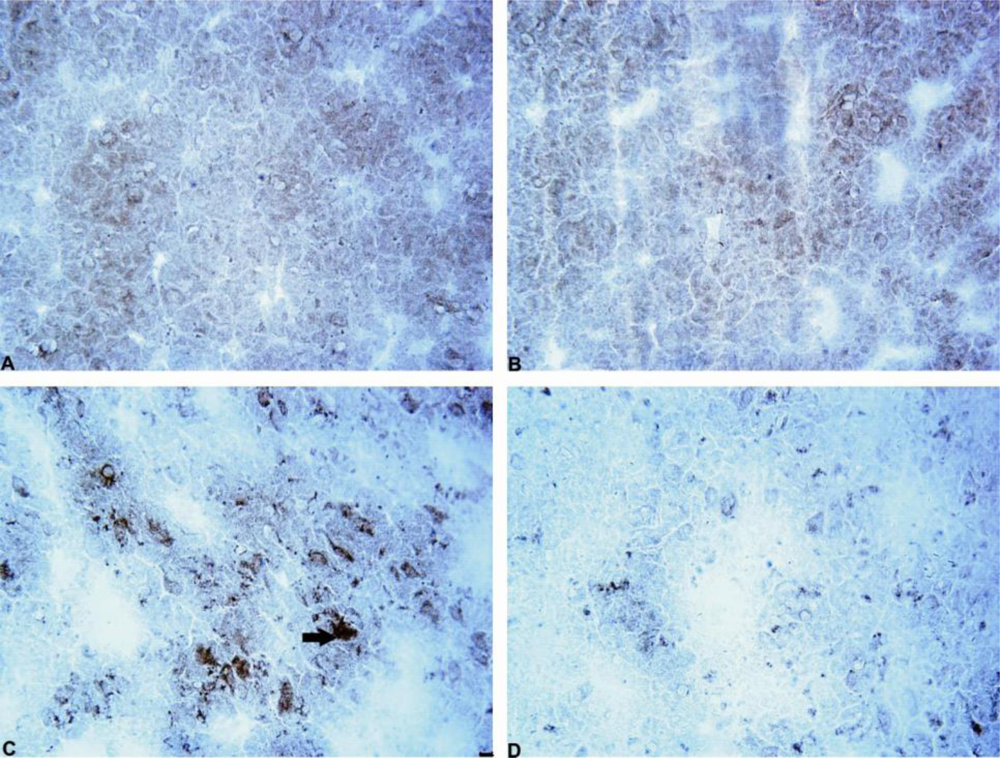
Our results demonstrated the ability of mildronate to completely prevent (to control level) the 6-OHDA-induced formation of ubiquitin intracellular inclusions in both the rat striatum and SN. The increased level of unconjugated ubiquitin and the formation of intracellular ubiquitin inclusions in the 6-OHDA dopamine-depleted striatum observed in the present study are consistent with the findings presented in the literature [
43]. The effect of mildronate leads us to suggest that it is capable of regulating the ubiquitin proteasome pathway, probably by activating the proteasome and/or enhancing ubiquitination and thereby promoting protein degradation. An explanation of this effect may be the ability of mildronate to protect cells at the level of the mitochondria [
38] because disturbances in ubiquitin production depend largely on oxidative stress-induced impairments in glucolysis and mitochondrial respiration [
44]. The central role of the ubiquitin-proteasome system in regulating the degradation of cellular proteins under different physiological conditions has been increasingly recognized.
The accumulation of misfolded proteins in the cells is involved in the pathogenesis of many neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD) [
19,
20,
43].
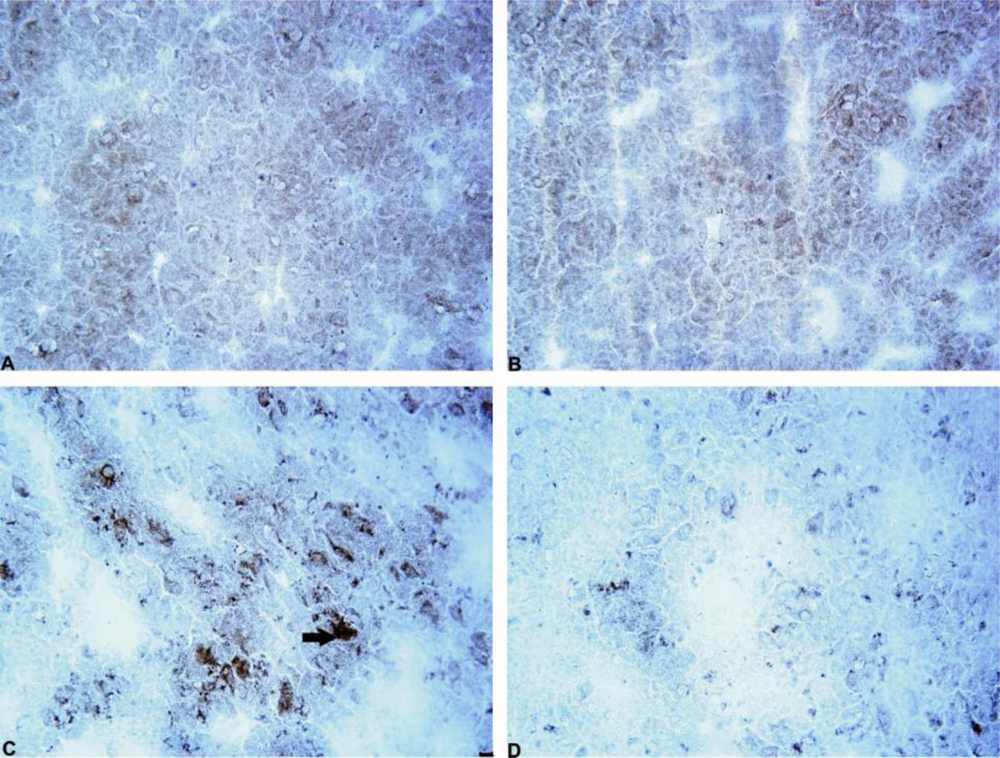
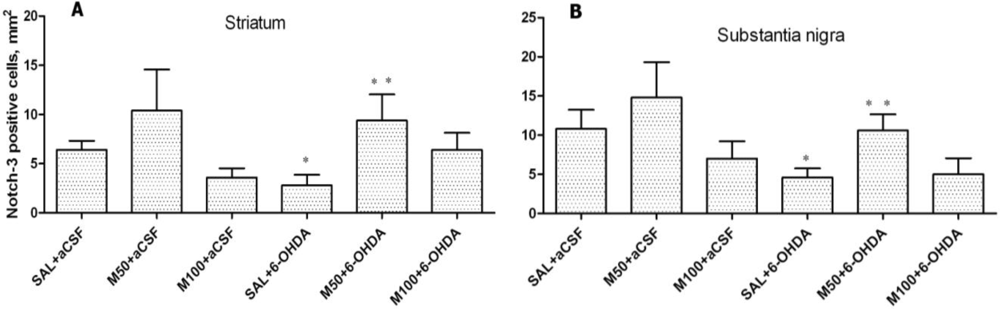
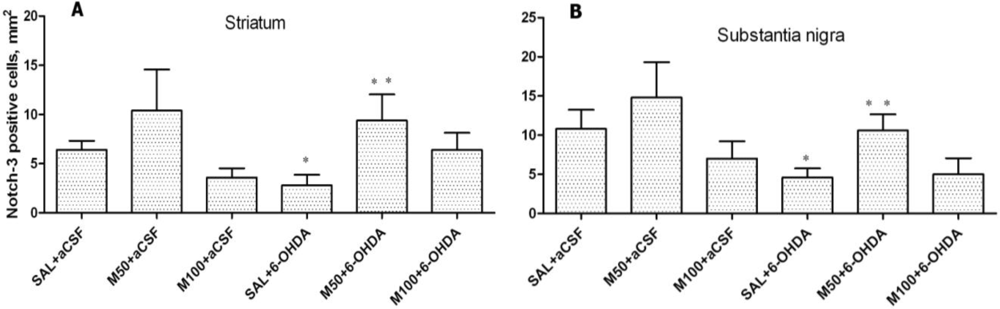
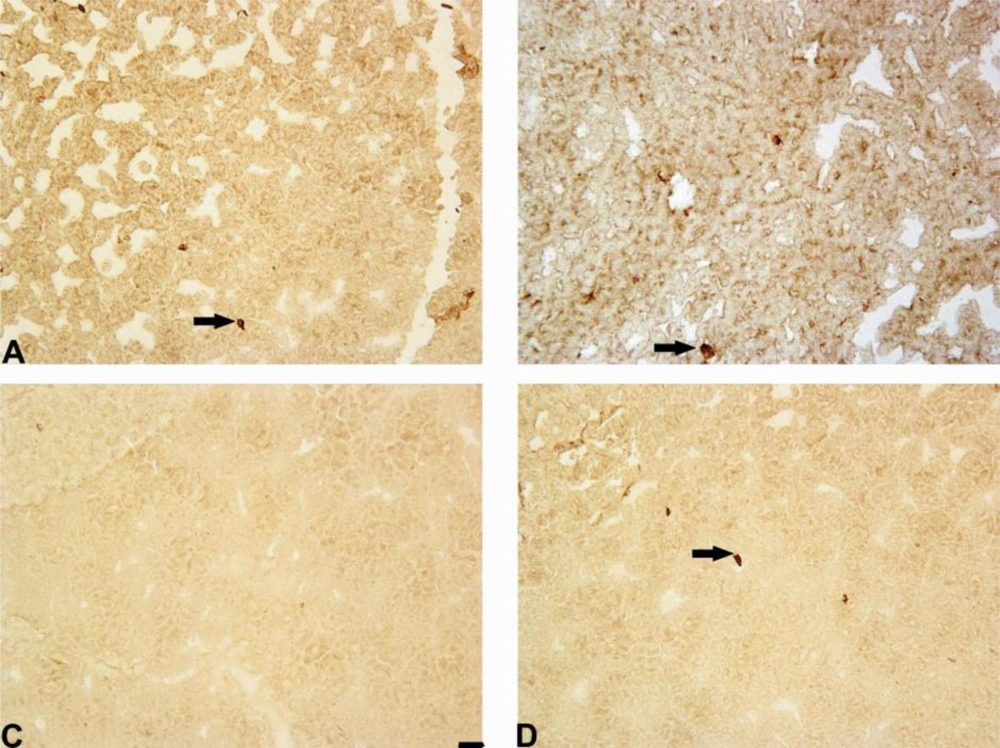
Our results have demonstrated that 6-OHDA decreased the number of Notch-3 positive cells in rat striatum and substantia nigra. However, mildronate at a dose of 50 mg/kg coadministrated with 6-OHDA increased the number of Notch-3-positive cells close to control values.
The Notch family of receptors and ligands plays an important role in the determination of cell fate, vasculogenesis and organogenesis, and the activation of Notch-1, 2 and 3 receptors has multiple roles during CNS development, particularly during gliogenesis [
45]. Furthermore, Notch-3 is a marker of progenitor cells and appears to couple the Notch pathway to the regulation of cell growth [
46]. The presence of endogenous progenitor cells in the adult mammalian brain presents an attractive alternative to the available therapeutic options for the treatment of PD [
32]. To date, we have not found any data showing Notch-3 expression in PD or PD animal models. The effect of mildronate on Notch-3 expression is difficult to explain. However, we think that it is an interesting phenomenon and suggest that mildronate stimulates the adult striatal progenitor cell population.
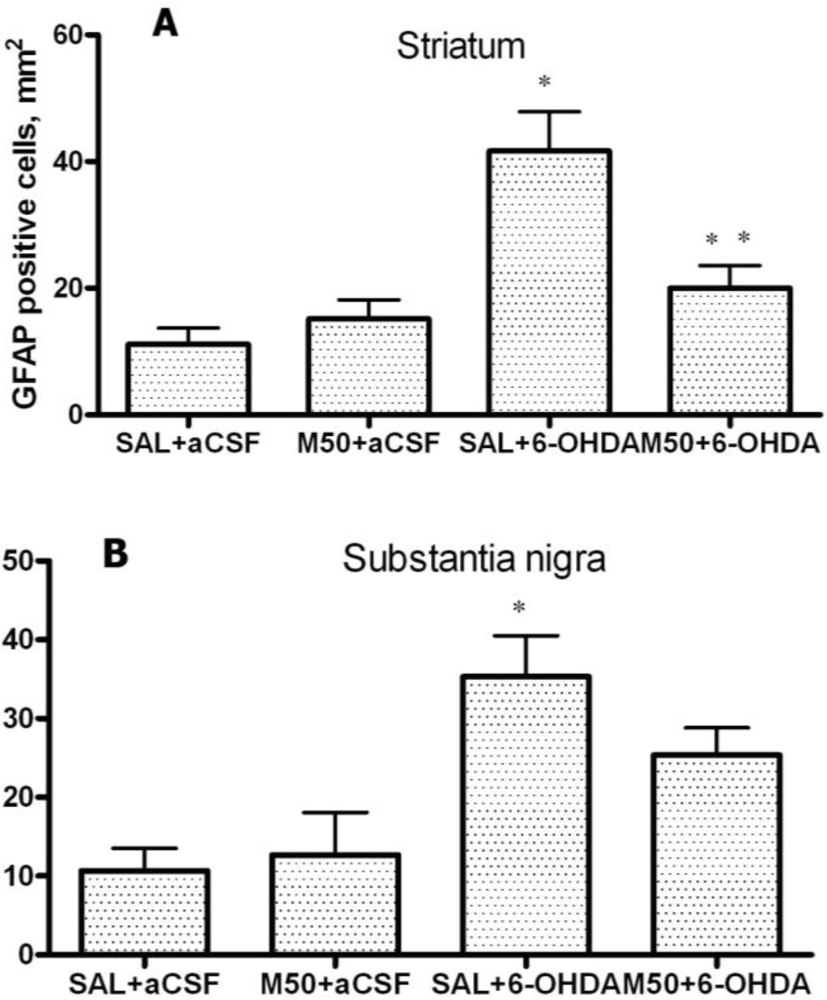
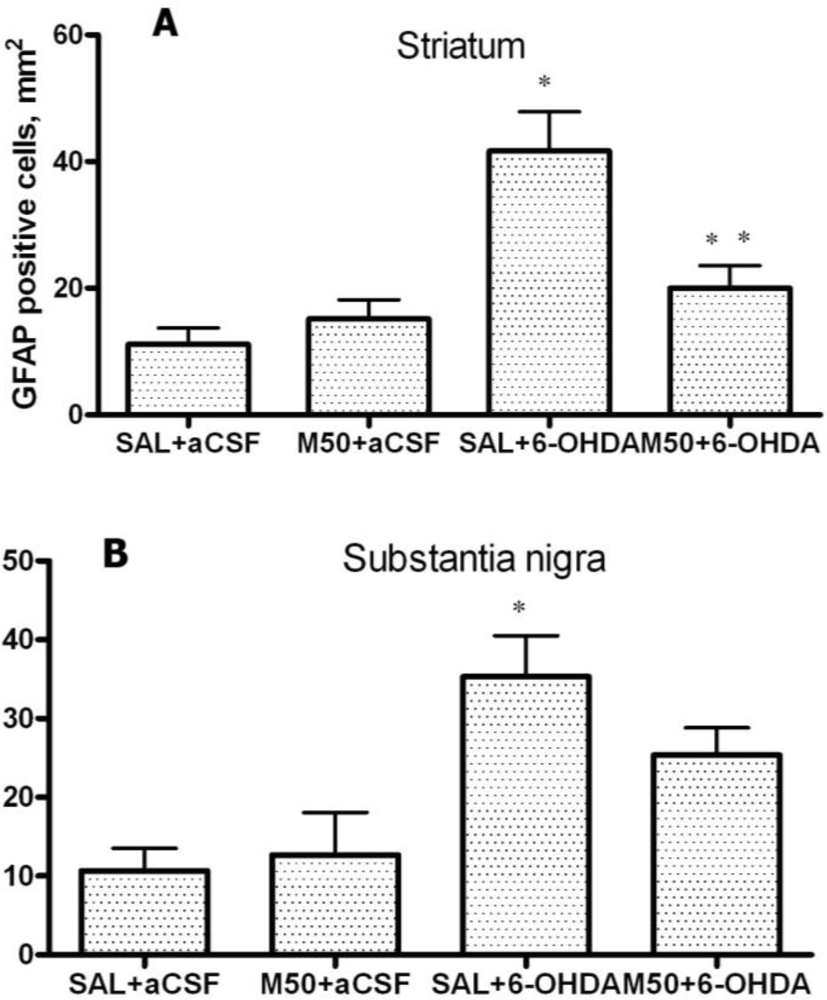
In addition, we demonstrated that 6-OHDA increased the number of GFAP-positive astrocytes in striatum and SN. This finding is consistent with previous data that demonstrated an increase in the levels of GFAP in 6-OHDA-lesioned structures [
32,
46]. The administration of mildronate at a dose of 50 mg/kg decreased the number of GFAP-positive astrocytes to a level similar to that detected in the control. We consider this phenomenon to be a positive effect of mildronate because it coincided with the normalization of TH expression, which affects cell survival. A similar normalization effect was observed in studies investigating the novel anti-parkinsonian agent zonisamide, which was found to attenuate MPTP-induced neurotoxicity in mice [
47].
There are different hypotheses concerning the mechanisms underlying reactive astrogliosis [
48,
49]. On the one hand, activated astrocytes may stimulate microglial cells, which induce dopamine cell sprouting via the synthesis of neurotrophic factors [
50]. Therefore, astrocytes can act as stem cells during adult neurogenesis [
51]. On the other hand, sustained inflammatory astrogliosis may have deteriorating effects after brain injuries and in neurodegenerative and demyelinating diseases [
51].
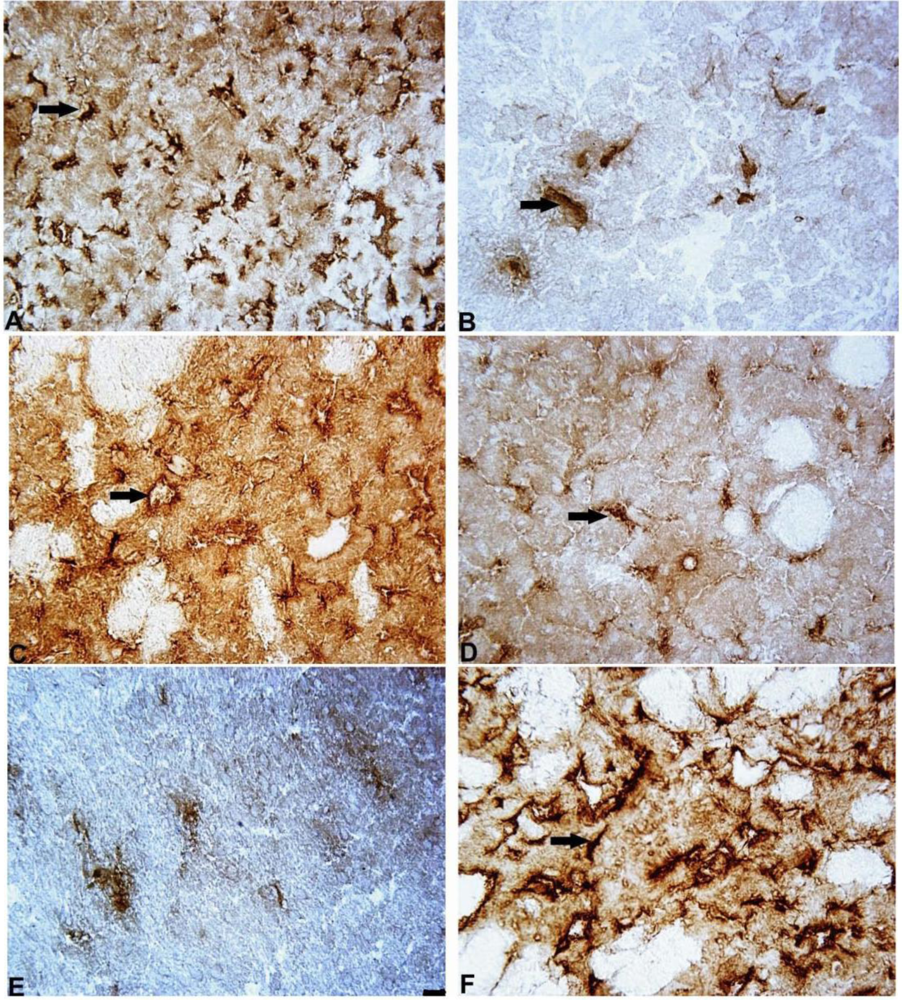
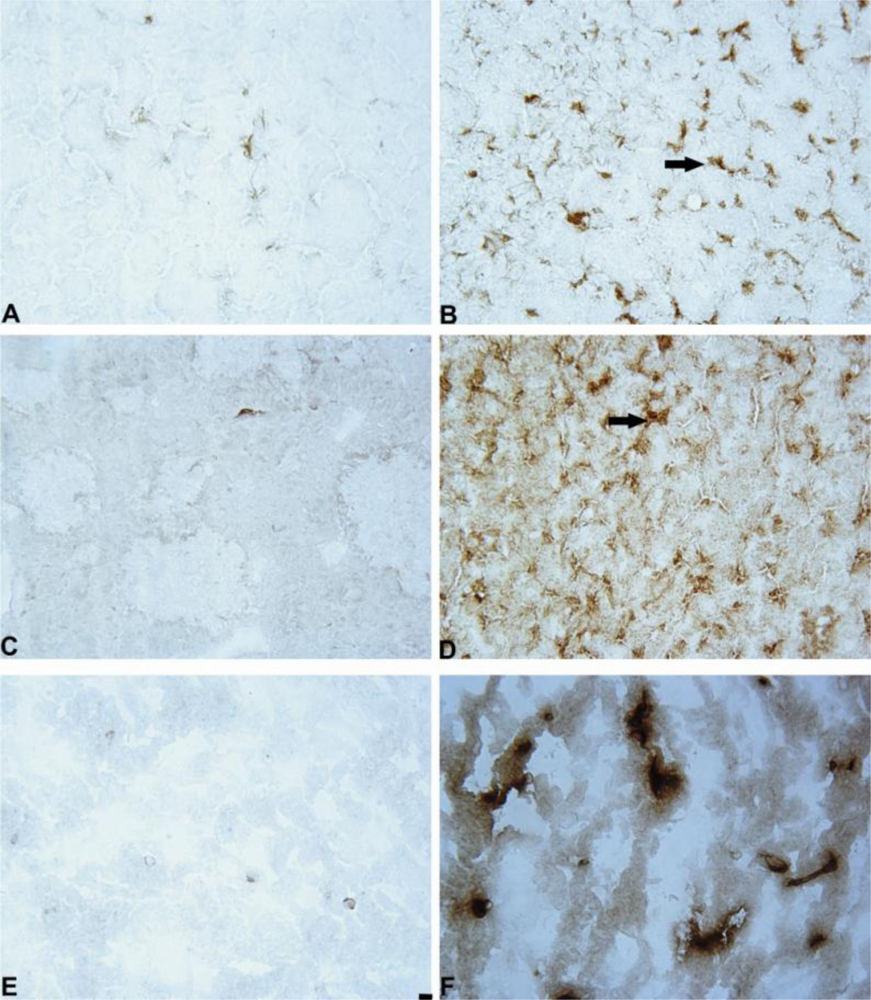
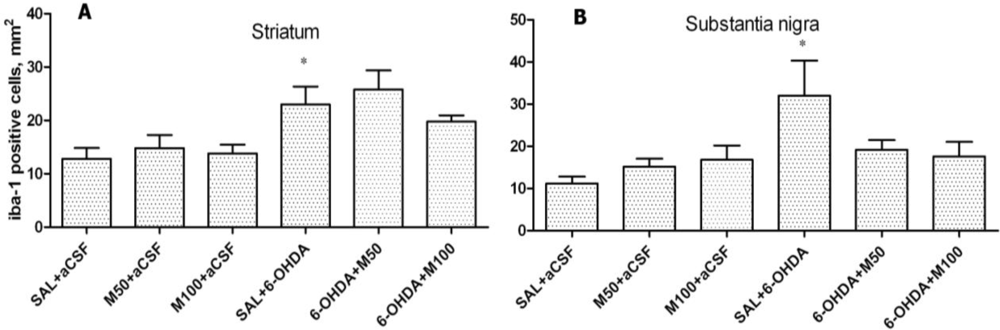
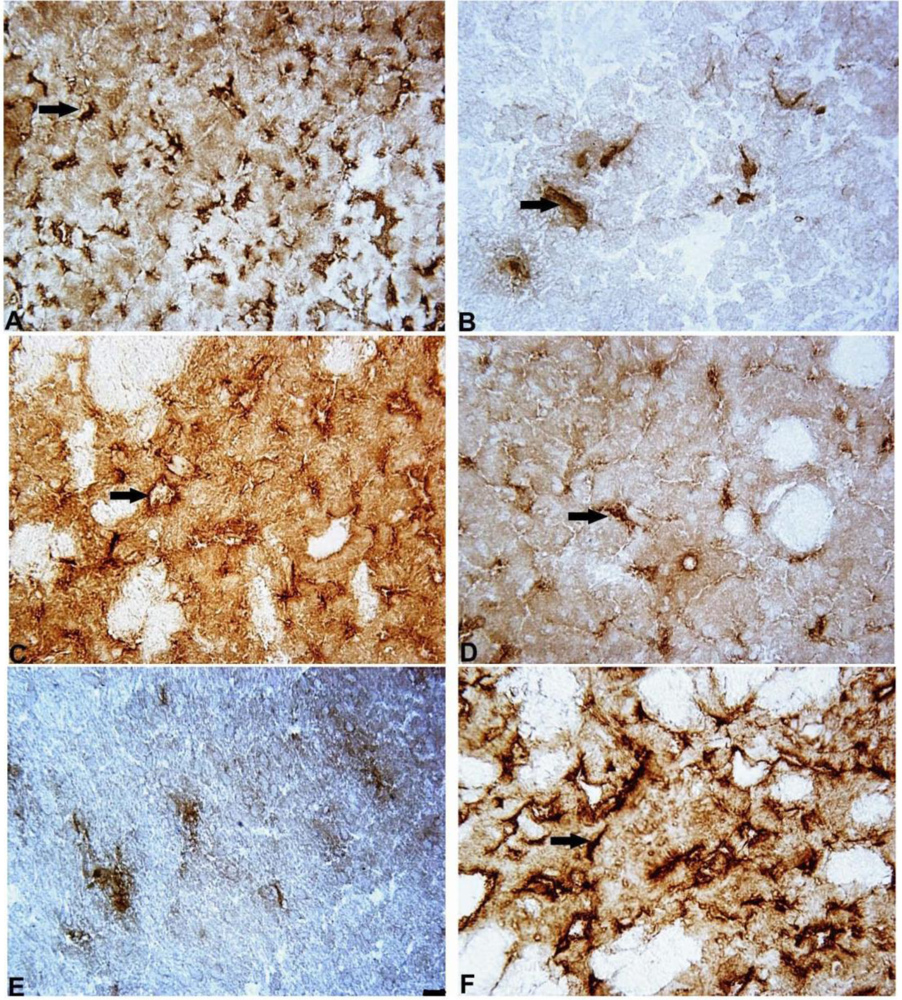
In the present study, we also assessed the microglial marker IBA-1, which is specifically expressed in macrophages/microglia and is upregulated when these cells are activated [
52]. We found that the expression of IBA-1 was increased by two-fold in the 6-OHDA-lesioned striatum and by 2.5-fold in the SN in comparison to the control group. These findings are consistent with the results described in the literature, which show that 6-OHDA causes neuroinflammation and microglial activation [
21] by increasing the expression of proinflammatory cytokines [
52]. Interestingly, in the 6-OHDA-lesioned SN, microglial activation precedes dopamine neuronal loss [
21,
22]. Microglial activation is important because reactive microglia may have a destructive influence on neurons. Conversely, activated microglia may stimulate the production of substances with neurotrophic properties [
50]. In the present study, mildronate did not affect the level of IBA-1 in the 6-OHDA-lesioned striatum, but in the SN, it showed a tendency to decrease the expression of IBA-1. Because IBA-1 activation coincided with an increase in TH and Notch-3 expression in the presence of mildronate, microglial activation may have a beneficial role in cell survival.
Finally, in the 6-OHDA-lesioned striatum and SN, we detected a considerable increase in the expression of the inflammatory marker iNOS, which indicated a manifestation of neuroinflammatory processes. Similarly, iNOS overexpression has been previously observed in the 6-OHDA model [
53], which indicates that iNOS induction plays an important role in the initial phase of neurodegeneration. The administration of mildronate at a dose of 50 mg/kg decreased the expression of iNOS in the SN by approximately two-fold, which indicates that mildronate possesses anti-inflammatory activity.
In summary, our data suggest that mildronate is capable of regulating many endogenous molecules that are involved in cell survival processes. The most important findings in the present study include the complete prevention of the loss of TH in the 6-OHDA-lesioned striatum and the decreased number of ubiquitin-positive intracellular inclusions. Furthermore, mildronate decreased GFAP and iNOS expression but stimulated Notch-3 expression. These data indicate that mildronate controls an array of cell proteins that participate in multi-faceted homeostatic mechanisms and provide neuroprotection in PD.
4. Experimental Section
4.1. Materials and Methods
Animals. Male Wistar rats were obtained from the Laboratory of Experimental Animals, Riga Stradins University, Riga, Latvia. Each animal weighed 230–250 g at the beginning of each experiment. All of the experimental procedures were performed in accordance with the guidelines of Directive 86/609/EEC “European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes” (1986) and were approved by the Animal Ethics Committee of the Food and Veterinary Service (Riga, Latvia).
Drugs. Mildronate [3-(2,2,2-trimethylhydrazinium) propionate dihydrate] was obtained from the Joint Stock Company “Grindex” (Riga, Latvia), dissolved in saline and prepared as a 2% stock solution. The 6-hydroxydopamine (6-OHDA) was obtained from Sigma-Aldrich (St. Louis, MO, USA), dissolved in 0.2% ascorbic acid solution and prepared as a stock solution at 20 μg/3 μL. This solution was made fresh and protected from light exposure. Apomorphine from Sigma-Aldrich (St. Louis, MO, USA) was dissolved in 0.1% ascorbic acid solution. Ketamine, xylazine and imipramine were purchased from Sigma-Aldrich (St. Louis, MO, USA).
The following antibodies were used: rabbit polyclonal IBA-1 (Wako Chemicals, USA); rabbit polyclonal TH (Millipore, USA); rabbit polyclonal Notch-3 (Santa Cruz Biotechnology, USA); rabbit polyclonal ubiquitin (Santa Cruz Biotechnology, USA); rabbit polyclonal GFAP (DAKO, Denmark); and rabbit polyclonal iNOS (Abcam, UK).
The EnVision detection kit, peroxidase-conjugated polyclonal goat anti-rabbit IgG and 3,3-diaminobenzidine (DAB) were purchased from DAKO, Denmark.
4.2. Experimental Design
The rats were adapted to the experimental conditions and divided into six groups (eight animals per group). Mildronate or saline (control) in a volume of 1 mL/kg was administered intraperitoneally (i.p.) every day for 14 days at doses of 50 or 100 mg/kg. On day 15, the neurotoxin 6-OHDA was administered into the right corpus striatum at a concentration of 20 μg/3 μL. The control group received artificial cerebrospinal fluid (aCSF).
Groups:
Group 1: Saline (i.p.) for two weeks followed by aCSF (intrastriatal)(corpus striatum).
Group 2: Mildronate, 50 mg/kg i.p. for two weeks followed by aCSF (intrastriatal).
Group 3: Mildronate, 100 mg/kg i.p. for two weeks followed by aCSF (intrastriatal).
Group 4: Saline i.p. for two weeks followed by 6-OHDA (intrastriatal).
Group 5: Mildronate, 50 mg/kg i.p. for two weeks followed by 6-OHDA (intrastriatal).
Group 6: Mildronate, 100 mg/kg i.p. for two weeks followed by 6-OHDA (intrastriatal).
4.3. Surgical Procedures
After the intraperitoneal administration of mildronate or saline for two weeks, the rats were anesthetized (ketamine 75 mg/kg + xylazine 10 mg/kg) and placed in a stereotaxic frame (Stoelting Inc., USA). Thirty minutes before the induction of general anesthesia, the rats received imipramine (at a dose of 20 mg/kg) to protect adrenergic neurons against the development of 6-OHDA-induced lesions.
Anesthetized rats received 20 μg/3 μL 6-OHDA solution or aCSF in the right striatum using a Stoelting microinjector. The injection rate was 1 μL/min, and the cannula was maintained in the delivery position for an additional 3 min prior to its slow retraction. Coordinates (according to Paxinos & Watson): AP + 1.2; LM + 2.5; DV − 5.0 mm from the bregma.
4.4. Rotational Behavior
On days 14, 21 and 28 after surgery and the administration of 6-OHDA, 0.2 mg/kg apomorphine (dopamine receptor agonist) was injected subcutaneously, and the number of asymmetric (contralateral) rotations for each animal was monitored over 30 min.
4.5. Brain Tissue Processing
After termination of the rotational behavior test (on day 28), the rats were anesthetized with ketamine (150 mg/kg) and perfused through the ascending aorta with 50 mL of isotonic saline followed by 250 mL of 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The brains were removed and fixed at −80 °C.
The brain tissue was cut into 10-μm-thick sections using a cryostat at −20 °C (Leica CM1850, Leica Microsystems, Germany), and 24 sections each from the corpus striatum (striatum) and substantia nigra (s. nigra) were obtained.
The sections were transferred to polylysine-coated slides (3 sections on each slide). The slides were air-dried for 15 minutes and then immersed in ice-cold acetone for 15 min and air-dried for 2 hours. The slides were wrapped in aluminum foil and stored at −20 °C.
4.6. Immunohistochemical Examination of the Brain Tissue
Ten-micron-thick tissue sections were stained to visualize the cells that were positive for TH, ubiquitin, Notch-3, IBA-1, GFAP and iNOS, according to a previously described immunohistochemical procedure [
40].
Briefly, endogenous peroxidase activity was blocked with 3.0% H2O2 for 10 min. Nonspecific primary antibody binding was blocked by incubating the slides with normal horse serum. These slides were incubated with rabbit polyclonal IBA-1 (Wako Chemicals, dilution at 1:500), rabbit polyclonal TH (Millipore, 1:400), or Notch-3 (Santa Cruz Biotechnology, 1:100), ubiquitin (1:100), or iNOS (Abcam, 1:100) overnight at 4 °C. The slides were incubated with rabbit polyclonal GFAP (DAKO, 1:500) for one hour at room temperature.
The rabbit polyclonal ubiquitin antibody was obtained from Santa Cruz Biotechnology (sc9133, clone FL-76). The epitope of the ubiquitin antibody corresponded to amino acids 1–76 and represented a full-length ubiquitin molecule of human origin. This antibody labels human, mouse and rat ubiquitin and polyubiquitin.
For TH, GFAP and iNOS immunostaining, the detection of bound antibody was performed using EnVision reagent (Dako, Denmark). To detect the expression of Notch-3, IBA-1 and ubiquitin, the slides were incubated with peroxidase-conjugated polyclonal goat anti-rabbit IgG for one hour.
The immunoperoxidase colour reaction was developed by incubating the slides with diaminobenzidine (5 min). A negative control without primary antibody was included in each experiment.
4.7. Imaging and Quantitation of Fibers and Cells
Quantitation of cells in the striatum. Tyrosine hydroxylase-immunopositive fibers were counted bilaterally in six independent sections each from all of the experimental groups. The total number of TH-positive nerve fibers in the striatum was quantified using a ×40 objective. The region of interest was captured using a Motic digital camera (Motic, China) mounted on a microscope (Motic BA400) using Motic Image Advanced 3.2 software. Whole striatal sections were captured and analyzed. The results are expressed as fibers per square millimeter.
A similar approach was utilized to count the number of GFAP, iNOS and IBA-1-positive cells. In addition, the number of cells that contained ubiquitin-positive inclusions was determined.
Quantitation of cells in the substantia nigra. To quantify the number of tyrosine hydroxylase (TH)-positive neurons, the positive cells were counted using a Motic BA400 microscope connected to a camera at a magnification of ×40. TH-positive neurons were counted in both the ipsilateral and contralateral hemispheres of the brain in each of the stereotaxic regions of the SN and adjacent ventral tegmental area (VTA) area to ensure complete sampling of the entire structure. The stereotaxic region in each section was determined by examining the shape and distribution of TH-positive neurons at a low magnification (4×). The substantia nigra (SN) was delineated from the VTA by reference to the specific anatomical landmark of the third cranial nerve rootlet. To minimize bias, all of the operators were assessed for counting consistency at each stereotaxic level of the SN prior to performing the cell counting. Intact cells with a nucleus, clear cytoplasm and axonal processes were included in the cell counts, and cells that appeared deformed and that lacked clear axonal processes were excluded from the cell counts. For each animal, six consecutive sections were counted at each stereotaxic level, and the results were pooled to obtain a total mean cell count for the ipsilateral and contralateral hemispheres in each group. To ensure an unbiased cell count, all of the operators were blinded to the analyzed group.
A similar approach was utilized to count the numbers of GFAP, iNOS and IBA-1-positive cells. In addition, the number of cells that contained ubiquitin-positive inclusions was counted.
4.8. Statistics
For the statistical analysis, GraphPad Prism 5 software was used. The results are expressed as the mean ± SEM, and the level of significance was p < 0.05 (unpaired t-test).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}