Effective Population Size, Gene Flow, and Species Status in a Narrow Endemic Sunflower, Helianthus neglectus, Compared to Its Widespread Sister Species, H. petiolaris

Abstract
:
1. Introduction
2. Results and Discussion
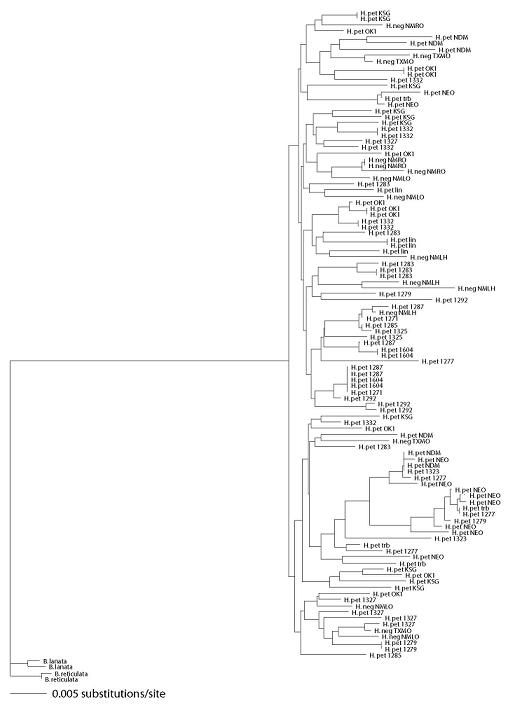
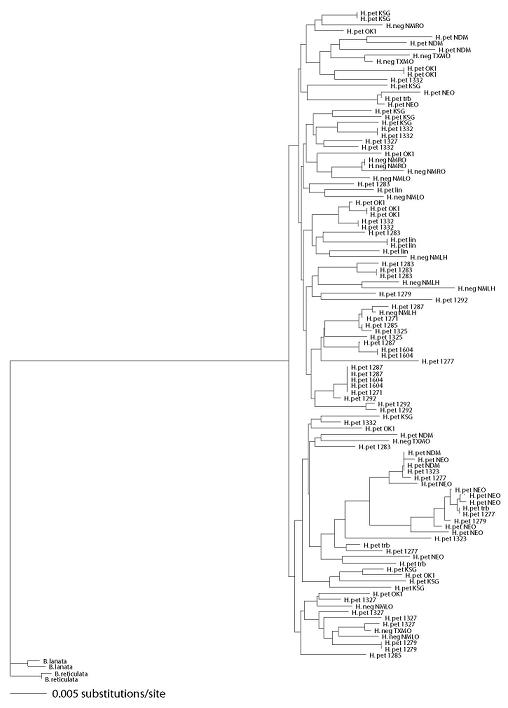
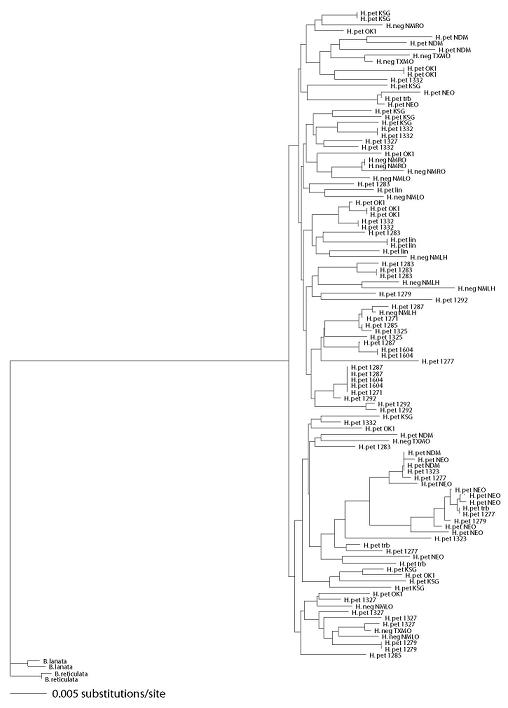
2.1. Genetic Diversity and Differentiation
2.2. Effective Population Sizes and Gene Flow Rates
2.3. Species Status of H. neglectus
3. Experimental Section
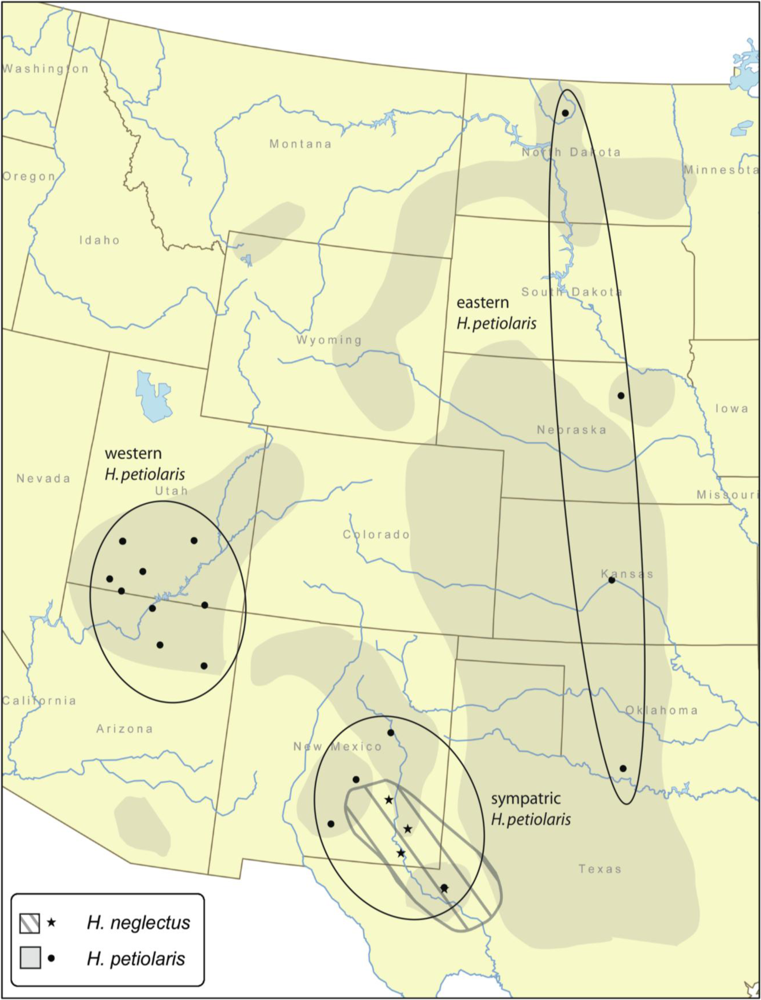
3.1. Sampling and Molecular Methods
3.2. Data Analysis
4. Conclusions
Acknowledgments
References and Notes
- De Queiroz, K. Species concepts and species delimitation. Syst. Biol 2007, 56, 879–886. [Google Scholar]
- Wiens, JJ; Penkrot, TA. Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloporus). Syst. Biol 2002, 51, 69–91. [Google Scholar]
- Young, ND. Pacific Coast Iris species delimitation using three species definitions: Biological, phylogenetic and genealogical. Biol. J. Linn. Soc 1998, 63, 99–120. [Google Scholar]
- Highton, R. Speciation in eastern North American salamanders of the genus Plethodon. Annu. Rev. Ecol. Syst 1995, 26, 579–600. [Google Scholar]
- Marthinsen, G; Wennerberg, L; Lifjeld, JT. Low support for separate species within the redpoll complex (Carduelis flammea-hornemanni-cabaret) from analyses of mtDNA and microsatellite markers. Mol. Phylogenet. Evol 2008, 47, 1005–1017. [Google Scholar]
- Becker, U; Dostal, P; Jorritsma-Wienk, LD; Matthies, D. The spatial scale of adaptive population differentiation in a wide-spread, well-dispersed plant species. Oikos 2008, 117, 1865–1873. [Google Scholar]
- Crispo, E. Modifying effects of phenotypic plasticity on interactions among natural selection, adaptation and gene flow. J. Evol. Biol 2008, 21, 1460–1469. [Google Scholar]
- Helsen, P; Browne, RA; Anderson, DJ; Verdyck, P; Dongen, S. Galapagos' Opuntia (prickly pear) cacti: extensive morphological diversity, low genetic variability. Biol. J. Linn. Soc 2009, 96, 451–461. [Google Scholar]
- Heiser, CB; Smith, DM; Clevenger, S; Martin, WC. The North American sunflowers (Helianthus). Mem. Torrey Bot. Club 1969, 22, 1–218. [Google Scholar]
- Rogers, CE; Thompson, TE; Seiler, GJ. Sunflower Species of the United States; National Sunflower Association: Bismarck, ND, USA, 1982; p. 75. [Google Scholar]
- Rieseberg, LH; Carter, R; Zona, S. Molecular tests of the hypothesized hybrid origin of two diploid Helianthus species (Asteraceae). Evolution 1990, 44, 1498–1511. [Google Scholar]
- Chandler, JM; Jan, C-C; Beard, BH. Chromosomal differentiation among the annual Helianthus species. Syst. Bot 1986, 11, 354–371. [Google Scholar]
- Heiser, CB. Three new annual sunflowers (Helianthus) from the southwestern United States. Rhodora 1958, 60, 272–283. [Google Scholar]
- Rieseberg, LH. Crossing relationships among ancient and experimental sunflower hybrid lineages. Evolution 2000, 54, 859–865. [Google Scholar]
- Rieseberg, LH; Carter, R; Zona, S. Molecular tests of the hypothesized hybrid origin of 2 diploid Helianthus species (Asteraceae). Evolution 1990, 44, 1498–1511. [Google Scholar]
- Wakeley, J; Hey, J. Estimating ancestral population parameters. Genetics 1997, 145, 847–855. [Google Scholar]
- Fu, YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Strasburg, JL; Rieseberg, LH. Molecular demographic history of the annual sunflowers Helianthus annuus and H. petiolaris – large effective population sizes and rates of long-term gene flow. Evolution 2008, 62, 1936–1950. [Google Scholar]
- Howard, DJ; Berlocher, SH. Endless Forms: Species and Speciation; Oxford University Press: New York, NY, USA, 1998; p. 470. [Google Scholar]
- Coyne, JA; Orr, HA. Speciation; Sinauer Associates: Sunderland, MA, USA, 2004. [Google Scholar]
- Templeton, AR. Using phylogeographic analyses of gene trees to test species status and processes. Mol. Ecol 2001, 10, 779–791. [Google Scholar]
- Alstrom, P; Rasmussen, PC; Olsson, U; Sundberg, P. Species delimitation based on multiple criteria: the spotted bush warbler Bradypterus thoracicus complex (Aves: Megaluridae). Zool. J. Linn. Soc 2008, 154, 291–307. [Google Scholar]
- Garcia-Paris, M; Buchholz, DR; Parra-Olea, G. Phylogenetic relationships of Pelobatoidea re-examined using mtDNA. Mol. Phylogenet. Evol 2003, 28, 12–23. [Google Scholar]
- Pfeil, BE; Brubaker, CL; Craven, LA; Crisp, MD. Phylogeny of Hibiscus and the tribe Hibisceae (Malvaceae) using chloroplast DNA sequences of ndhF and the rpl16 intron. Syst. Bot 2002, 27, 333–350. [Google Scholar]
- Cole, CT. Genetic variation in rare and common plants. Annu. Rev. Ecol. Evol. Syst 2003, 34, 213–237. [Google Scholar]
- Buerkle, CA; Morris, RJ; Asmussen, MA; Rieseberg, LH. The likelihood of homoploid hybrid speciation. Heredity 2000, 84, 441–451. [Google Scholar]
- Kane, NC; King, MG; Barker, MS; Raduski, A; Karrenberg, S; Yatabe, Y; Knapp, SJ; Rieseberg, LH. Comparative genomic and population genetic analyses indicate highly porous genomes and high levels of gene flow between divergent Helianthus species. Evolution 2009, 63, 2061–2075. [Google Scholar]
- Schwarzbach, AE; Rieseberg, LH. Likely multiple origins of a diploid hybrid sunflower species. Mol. Ecol 2002, 11, 1703–1715. [Google Scholar]
- Gross, BL; Schwarzbach, AE; Rieseberg, LH. Origin(s) of the diploid hybrid species Helianthus deserticola (Asteraceae). Am. J. Bot 2003, 90, 1708–1719. [Google Scholar]
- Welch, ME; Rieseberg, LH. Patterns of genetic variation suggest a single, ancient origin for the diploid hybrid species Helianthus paradoxus. Evolution 2002, 56, 2126–2137. [Google Scholar]
- Beerli, P. Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 2006, 22, 341–345. [Google Scholar]
- Heiser, CB. Morphological and cytological variation in Helianthus petiolaris with notes on related species. Evolution 1961, 15, 247–258. [Google Scholar]
- Yatabe, Y; Kane, NC; Scotti-Saintagne, C; Rieseberg, LH. Rampant gene exchange across a strong reproductive barrier between the annual sunflowers, Helianthus annuus and H. petiolaris. Genetics 2007, 175, 1883–1893. [Google Scholar]
- Strasburg, JL; Scotti-Saintagne, C; Scotti, I; Lai, Z; Rieseberg, LH. Genomic patterns of adaptive divergence between chromosomally differentiated sunflower species. Mol. Biol. Evol 2009, 26, 1341–1355. [Google Scholar]
- Burke, JM; Lai, Z; Salmaso, M; Nakazato, T; Tang, SX; Heesacker, A; Knapp, SJ; Rieseberg, LH. Comparative mapping and rapid karyotypic evolution in the genus Helianthus. Genetics 2004, 167, 449–457. [Google Scholar]
- Templeton, AR. Population Genetics and Microevolutionary Theory; Wiley-Liss: Hoboken, NJ, USA, 2006. [Google Scholar]
- Schilling, EE; Paner, JL. A revised classification of subtribe Helianthinae (Asteraceae: Heliantheae). I. Basal lineages. Bot. J. Linn. Soc 2002, 140, 65–76. [Google Scholar]
- Jukes, TH; Cantor, CR. Evolution of protein molecules. In Mammalian Protein Metabolism; Munro, HN, Ed.; Academic Press: New York, NY, USA, 1969; pp. 21–132. [Google Scholar]
- Watterson, GA. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol 1975, 7, 256–276. [Google Scholar]
- Rozas, J; Sanchez-DelBarrio, JC; Messeguer, X; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar]
- Hey, J; Wakeley, J. A coalescent estimator of the population recombination rate. Genetics 1997, 145, 833–846. [Google Scholar]
- Swofford, DL. PAUP* Phylogenetic Analysis Using Parsimony (*and Other Methods); , Beta Version 40b2; Sinauer: Sunderland, MA, USA, 1999. [Google Scholar]
- Excoffier, LGL; Schneider, S. Arlequin ver. 3.0: An Integrated software package for population genetics data analysis. Evol. Bioinf. Online 2005, 1, 47–50. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Hudson, RR; Kreitman, M; Aguade, M. A test of neutral molecular evolution based on nucleotide data. Genetics 1987, 116, 153–159. [Google Scholar]
- Wright, SI; Charlesworth, B. The HKA test revisited: A maximum-likelihood-ratio test of the standard neutral model. Genetics 2004, 168, 1071–1076. [Google Scholar]
- Beerli, P. Migrate version 3.0–A Maximum Likelihood and Bayesian Estimator of Gene Flow Using the Coalescent. 2008.
- Hudson, RR; Kaplan, NL. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar]
- Hey, J; Nielsen, R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl. Acad. Sci. USA 2007, 104, 2785–2790. [Google Scholar]
- Hey, J. Isolation with migreation models for more than two populations. Mol Biol Evol 2009. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| H. petiolaris | H. neglectus | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Linkage group | Aligned size (bp) | Protein homology (based on closest Arabidopsis thaliana BLAST hit) | No. of seqs. | π (silent) | θ (silent) | No. of seqs. | π (silent) | θ (silent) | % Gross divergence | % Net divergence |
| (2213) | 6 | 688 | Scarecrow transcription factor family protein | 32 | 0.019 | 0.024 | 16 | 0.017 | 0.022 | 2.66 | 0.46 |
| JLS244 | 9 | 571 | Proton-dependent oligopeptide transport (POT) family protein | 30 | 0.024 | 0.036 | 16 | 0.030 | 0.037 | 1.76 | 0.12 |
| JLS720a | 16 | 687 | Cellulose synthase-related protein | 32 | 0.028 | 0.037 | 2 | 0.040 | 0.039 | 1.93 | 0.00 |
| JLS810R1 | 13 | 1326 | Auxin efflux carrier protein family | 32 | 0.053 | 0.039 | 16 | 0.063 | 0.058 | 4.43 | 0.37 |
| JLS1040 | 10 | 659 | Serine/threonine phosphatase | 96 | 0.046 | 0.063 | 16 | 0.045 | 0.062 | 2.43 | 0.07 |
| JLS1615 | 14 | 575 | Unknown protein | 96 | 0.044 | 0.082 | 16 | 0.051 | 0.060 | 1.14 | 0.06 |
| JLS1747R2 | 17 | 789 | Amino acid permease family protein | 90 | 0.062 | 0.093 | 16 | 0.061 | 0.076 | 2.47 | 0.04 |
| JLS1953 | 17 | 454 | Galacturonosyltransferase 4 | 32 | 0.016 | 0.024 | 16 | 0.008 | 0.011 | 1.19 | 0.05 |
| JLS2899 | 14 | 776 | Unknown protein | 32 | 0.032 | 0.057 | 16 | 0.052 | 0.067 | 1.48 | 0.04 |
| Average | 725 | 0.036 | 0.051 | 0.041 | 0.048 | 2.17 | 0.14 | ||||
| Locus | Polymorphic in H. petiolaris only | Polymorphic in H. neglectus only | Polymorphic in both species | Fixed differences between species |
|---|---|---|---|---|
| (2213) | 33 | 30 | 15 | 0 |
| JLS244 | 24 | 22 | 13 | 0 |
| JLS720a | 40 | 8 | 10 | 0 |
| JLS810R1 | 27 | 44 | 39 | 0 |
| JLS1040 | 79 | 36 | 37 | 0 |
| JLS1615 | 47 | 8 | 19 | 0 |
| JLS1747R2 | 98 | 29 | 56 | 0 |
| JLS1953 | 32 | 5 | 9 | 0 |
| JLS2899 | 30 | 28 | 29 | 0 |
| Overall | 410 | 210 | 227 | 0 |
| H. petiolaris | H. neglectus | |||||||
|---|---|---|---|---|---|---|---|---|
| Tajima’s D | Fu’s Fs | Tajima’s D | Fu’s Fs | |||||
| Locus | D | p value | Fs | p value | D | p value | Fs | p value |
| (2213) | −0.491 | 0.353 | −14.547 | <0.001* | −0.738 | 0.234 | −2.918 | 0.052 |
| JLS244 | −0.857 | 0.214 | −24.231 | <0.001* | −0.893 | 0.218 | −9.227 | <0.001* |
| JLS720a | −1.008 | 0.166 | −23.772 | <0.001* | N/A | N/A | N/A | N/A |
| JLS810R1 | 1.423 | 0.952 | −6.959 | <0.008 | 0.344 | 0.702 | −5.905 | 0.006 |
| JLS1040 | −1.279 | 0.070 | −23.957 | <0.001* | −1.196 | 0.118 | −3.941 | 0.039 |
| JLS1615 | −1.721 | 0.017 | −25.183 | <0.001* | −0.932 | 0.196 | −11.118 | <0.001* |
| JLS1747R2 | −1.209 | 0.105 | −23.990 | 0.001* | −1.197 | 0.110 | −3.922 | 0.029 |
| JLS1953 | −1.326 | 0.074 | −24.396 | <0.001* | −0.878 | 0.205 | −6.512 | 0.005* |
| JLS2899 | −1.698 | 0.029 | −24.103 | <0.001* | −1.014 | 0.161 | −6.586 | 0.005* |
| Population | Mode | 2.5% | 97.5% |
|---|---|---|---|
| Eastern H. petiolaris | 2.42 × 105 | 0 | 4.87 × 105 |
| Western H. petiolaris | 2.92 × 105 | 4.74 × 104 | 5.33 × 105 |
| Sympatric H. petiolaris | 6.42 × 105 | 1.51 × 105 | 1.97 × 106 |
| H. neglectus | 2.29 × 106 | 8.45 × 105 | 9.20 × 106 |
| Gene Flow (Nem) | into EP | into WP | into SP | into NE |
|---|---|---|---|---|
| from EP | - | 0.01 (0–3.80) | 0.01 (0–4.98) | 0.01 (0–5.08) |
| from WP | 0.01 (0–3.80) | - | 0.01 (0–5.50) | 0.01 (0–4.83) |
| from SP | 0.01 (0–3.88) | 0.01 (0–3.78) | - | 0.01 (0–5.13) |
| from NE | 0.01 (0–3.90) | 0.01 (0–3.78) | 0.01 (0–4.70) | - |
| Species | Population | Location | Latitude (N) | Longitude (W) | No. of individuals |
|---|---|---|---|---|---|
| H. petiolaris | KSG1 | Great Bend, KS | 38.36 | 98.76 | 5 |
| NDM1 | Minot, ND | 48.30 | 100.71 | 5 | |
| NEO1 | O’Neill, NE | 42.29 | 98.63 | 5 | |
| OK11 | Cotton Co., OK | 34.33 | 98.40 | 5 | |
| Pet_12832 | Coconino Co., AZ | 36.04 | 111.19 | 3 | |
| Pet_12872 | Kane Co., UT | 37.24 | 112.86 | 2 | |
| Pet_16042 | Kane Co., UT | 37.03 | 112.48 | 2 | |
| Pet_12772 | Coconino Co., AZ | 36.78 | 111.55 | 2 | |
| Pet_12792 | Navajo Co., AZ | 35.75 | 109.91 | 2 | |
| Pet_12922 | Hanksville, UT | 38.35 | 110.69 | 2 | |
| Pet_12852 | Kane Co., UT | 37.52 | 111.98 | 1 | |
| Pet_13232 | San Juan Co., UT | 37.03 | 110.13 | 1 | |
| Pet_12712 | Iron Co., UT | 38.08 | 112.69 | 1 | |
| Pet_13252 | Iron Co., UT | 38.08 | 112.68 | 1 | |
| Pet_13273 | Puerto de Luna, NM | 34.82 | 104.63 | 3 | |
| Pet_13323 | Monahans, TX | 31.62 | 102.90 | 4 | |
| Pet_lin3 | Lincoln Co., NM | 33.95 | 105.41 | 2 | |
| Pet_trb3 | Tularosa Basin, NM | 32.90 | 105.96 | 2 | |
| H. neglectus | NMRO4 | Roswell, NM | 33.40 | 104.53 | 2 |
| NMLO4 | Loving, NM | 32.29 | 104.09 | 2 | |
| NMLH4 | Loco Hills, NM | 32.82 | 103.97 | 2 | |
| TXMO4 | Monahans, TX | 31.60 | 102.89 | 2 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Raduski, A.R.; Rieseberg, L.H.; Strasburg, J.L. Effective Population Size, Gene Flow, and Species Status in a Narrow Endemic Sunflower, Helianthus neglectus, Compared to Its Widespread Sister Species, H. petiolaris. Int. J. Mol. Sci. 2010, 11, 492-506. https://doi.org/10.3390/ijms11020492
Raduski AR, Rieseberg LH, Strasburg JL. Effective Population Size, Gene Flow, and Species Status in a Narrow Endemic Sunflower, Helianthus neglectus, Compared to Its Widespread Sister Species, H. petiolaris. International Journal of Molecular Sciences. 2010; 11(2):492-506. https://doi.org/10.3390/ijms11020492
Chicago/Turabian StyleRaduski, Andrew R., Loren H. Rieseberg, and Jared L. Strasburg. 2010. "Effective Population Size, Gene Flow, and Species Status in a Narrow Endemic Sunflower, Helianthus neglectus, Compared to Its Widespread Sister Species, H. petiolaris" International Journal of Molecular Sciences 11, no. 2: 492-506. https://doi.org/10.3390/ijms11020492
APA StyleRaduski, A. R., Rieseberg, L. H., & Strasburg, J. L. (2010). Effective Population Size, Gene Flow, and Species Status in a Narrow Endemic Sunflower, Helianthus neglectus, Compared to Its Widespread Sister Species, H. petiolaris. International Journal of Molecular Sciences, 11(2), 492-506. https://doi.org/10.3390/ijms11020492