Nitric Oxide: Perspectives and Emerging Studies of a Well Known Cytotoxin

Abstract
:
1. Background
2. GST-pi
3. Reactive Nitrogen Species
4. Epigenetics and NO•
4.1. DNA Methylation
4.2. MicroRNA
4.3. Histone Modifications
5. Biological Model System
6. Conclusion
References and Notes
- Furchgott, RF; Zawadzki, JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar]
- Stuehr, DJ; Marletta, MA. Mammalian nitrate biosynthesis: Mouse macrophages produce nitrite and nitrate in response to escherichia coli lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1985, 82, 7738–7742. [Google Scholar]
- Jacklet, JW. Nitric oxide signaling in invertebrates. Invert. Neurosci 1997, 3, 1–14. [Google Scholar]
- Wilson, ID; Neill, SJ; Hancock, JT. Nitric oxide synthesis and signalling in plants. Plant Cell Environ 2008, 31, 622–631. [Google Scholar]
- Illi, B; Colussi, C; Grasselli, A; Farsetti, A; Capogrossi, MC; Gaetano, C. No sparks off chromatin: Tales of a multifaceted epigenetic regulator. Pharmacol. Ther 2009, 123, 344–352. [Google Scholar]
- Bentz, BG; Simmons, RL; Haines, GK, III; Radosevich, JA. The yin and yang of nitric oxide: Reflections on the physiology and pathophysiology of no. Head Neck 2000, 22, 71–83. [Google Scholar]
- Mocellin, S; Bronte, V; Nitti, D. Nitric oxide, a double edged sword in cancer biology: Searching for therapeutic opportunities. Med. Res. Rev 2007, 27, 317–352. [Google Scholar]
- Stamler, JS. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 78, 931–936. [Google Scholar]
- Hibbs, JB; Vavrin, A; Taintor, RR. L-arginine is required for expression of the activated macrophage effector mechanism causing selective metabolic inhibition in target cells. J. Immunol 1987, 138, 550–565. [Google Scholar]
- Langrehr, JM; Hoffman, RA; Lancaster, JR, Jr; Simmons, RL. Nitric oxide--a new endogenous immunomodulator. Transplantation 1993, 55, 1205–1212. [Google Scholar]
- Garthwaite, J. Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci 2008, 27, 2783–2802. [Google Scholar]
- Chen, J; Zacharek, A; Zhang, C; Jiang, H; Li, Y; Roberts, C; Lu, M; Kapke, A; Chopp, M. Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J. Neurosci 2005, 25, 2366–2375. [Google Scholar]
- Goud, AP; Goud, PT; Diamond, MP; Abu-Soud, HM. Nitric oxide delays oocyte aging. Biochemistry 2005, 44, 11361–11368. [Google Scholar]
- Stamler, JS; Meissner, G. Physiology of nitric oxide in skeletal muscle. Physiol. Rev 2001, 81, 209–237. [Google Scholar]
- Teixeira, CC; Agoston, H; Beier, F. Nitric oxide, c-type natriuretic peptide and cgmp as regulators of endochondral ossification. Dev. Biol 2008, 319, 171–178. [Google Scholar]
- Hayashi, T; Yano, K; Matsui-Hirai, H; Yokoo, H; Hattori, Y; Iguchi, A. Nitric oxide and endothelial cellular senescence. Pharmacol. Ther 2008, 120, 333–339. [Google Scholar]
- Spinetti, G; Kraenkel, N; Emanueli, C; Madeddu, P. Diabetes and vessel wall remodelling: From mechanistic insights to regenerative therapies. Cardiovasc. Res 2008, 78, 265–273. [Google Scholar]
- Thomas, DD; Espey, MG; Ridnour, LA; Hofseth, LJ; Mancardi, D; Harris, CC; Wink, DA. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc. Natl. Acad. Sci. USA 2004, 101, 8894–8899. [Google Scholar]
- Ridnour, LA; Thomas, DD; Mancardi, D; Espey, MG; Miranda, KM; Paolocci, N; Feelisch, M; Fukuto, J; Wink, DA. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol. Chem 2004, 385, 1–10. [Google Scholar]
- Yu, Z; Kuncewicz, T; Dubinsky, WP; Kone, BC. Nitric oxide-dependent negative feedback of parp-1 trans-activation of the inducible nitric-oxide synthase gene. J. Biol. Chem 2006, 281, 9101–9109. [Google Scholar]
- Mocellin, S. Nitric oxide: Cancer target or anticancer agent? Curr. Cancer Drug Targets 2009, 9, 214–236. [Google Scholar]
- Ng, QS; Goh, V; Milner, J; Stratford, MR; Folkes, LK; Tozer, GM; Saunders, MI; Hoskin, PJ. Effect of nitric-oxide synthesis on tumour blood volume and vascular activity: A phase I study. Lancet Oncol 2007, 8, 111–118. [Google Scholar]
- Gratton, JP; Lin, MI; Yu, J; Weiss, ED; Jiang, ZL; Fairchild, TA; Iwakiri, Y; Groszmann, R; Claffey, KP; Cheng, YC; et al. Selective inhibition of tumor microvascular permeability by cavtratin blocks tumor progression in mice. Cancer Cell 2003, 4, 31–39. [Google Scholar]
- Murohara, T; Asahara, T; Silver, M; Bauters, C; Masuda, H; Kalka, C; Kearney, M; Chen, D; Symes, JF; Fishman, MC; et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Invest 1998, 101, 2567–2578. [Google Scholar]
- Papapetropoulos, A; Garcia-Cardena, G; Madri, JA; Sessa, WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest 1997, 100, 3131–3139. [Google Scholar]
- Crowell, JA; Steele, VE; Sigman, CC; Fay, JR. Is inducible nitric oxide synthase a target for chemoprevention? Mol. Cancer Ther 2003, 2, 815–823. [Google Scholar]
- Hofseth, LJ; Hussain, SP; Wogan, GN; Harris, CC. Nitric oxide in cancer and chemoprevention. Free Radic. Biol. Med 2003, 34, 955–968. [Google Scholar]
- Tozer, GM; Prise, VE; Chaplin, DJ. Inhibition of nitric oxide synthase induces a selective reduction in tumor blood flow that is reversible with l-arginine. Cancer Res 1997, 57, 948–955. [Google Scholar]
- Haynes, WG; Noon, JP; Walker, BR; Webb, DJ. Inhibition of nitric oxide synthesis increases blood pressure in healthy humans. J. Hypertens 1993, 11, 1375–1380. [Google Scholar]
- Lorente, JA; Landin, L; De Pablo, R; Renes, E; Liste, D. L-arginine pathway in the sepsis syndrome. Crit. Care Med 1993, 21, 1287–1295. [Google Scholar]
- Bentz, BG; Haines, GK, III; Radosevich, JA. Increased protein nitrosylation in head and neck squamous cell carcinoma. Head Neck 2000, 22, 64–70. [Google Scholar]
- Jenkins, DC; Charles, IG; Thomsen, LL; Moss, DW; Holmes, LS; Baylis, SA; Rhodes, P; Westmore, K; Emson, PC; Moncada, S. Roles of nitric oxide in tumor growth. Proc. Natl. Acad. Sci. USA 1995, 92, 4392–4396. [Google Scholar]
- Epperlein, MM; Nourooz-Zadeh, J; Noronha-Dutra, AA; Woolf, N. Nitric oxide in cigarette smoke as a mediator of oxidative damage. Int. J. Exp. Pathol 1996, 77, 197–200. [Google Scholar]
- Gaston, B; Drazen, JM; Loscalzo, J; Stamler, JS. The biology of nitrogen oxides in the airways. Am. J. Respir. Crit. Care Med 1994, 149, 538–551. [Google Scholar]
- Mirvish, SS. Role of n-nitroso compounds (noc) and n-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to noc. Cancer Lett 1995, 93, 17–48. [Google Scholar]
- Gibson, QH; Roughton, FJ. The determination of the velocity constants of four successive reactions of carbon monoxide with sheep haemoglobin. Proc Soc Ser B: Biol Sci Lond 1957, 206–224. [Google Scholar]
- Davis, KL; Martin, E; Turko, IV; Murad, F. Novel effects of nitric oxide. Annu. Rev. Pharmacol. Toxicol 2001, 41, 203–236. [Google Scholar]
- Souza, JM; Peluffo, G; Radi, R. Protein tyrosine nitration--functional alteration or just a biomarker? Free Radic. Biol. Med 2008, 45, 357–366. [Google Scholar]
- Lane, AP; Prazma, J; Baggett, HC; Rose, AS; Pillsbury, HC. Nitric oxide is a mediator of neurogenic vascular exudation in the nose. Otolaryngol. Head Neck. Surg 1997, 116, 294–300. [Google Scholar]
- Michel, O; Bloch, W; Rocker, J. Nos-mapping of human nasal mucosa under physiologic and pathophysiological conditions. In Proceedings of the Fourth International Meeting on the Biology of Nitric Oxide; Moncada, S, Stamler, J, Gross, S, Higgs, EA, Eds.; Portland Press: London, UK, 1996; 230. [Google Scholar]
- Eaton, DL; Bammler, TK. Concise review of the glutathione s-transferases and their significance to toxicology. Toxicol. Sci 1999, 49, 156–164. [Google Scholar]
- Hanna, E; MacLeod, S; Vural, E; Lang, N. Genetic deletions of glutathione-s-transferase as a risk factor in squamous cell carcinoma of the larynx: A preliminary report. Am. J. Otolaryngol 2001, 22, 121–123. [Google Scholar]
- Ketterer, B. Protective role of glutathione and glutathione transferases in mutagenesis and carcinogenesis. Mutat. Res 1988, 202, 343–361. [Google Scholar]
- Mulder, TP; Manni, JJ; Roelofs, HM; Peters, WH; Wiersma, A. Glutatione s-transferases and glutathione in human head and neck cancer. Carcinogenesis 1995, 16, 619–624. [Google Scholar]
- McIlwain, CC; Townsend, DM; Tew, KD. Glutathione s-transferase polymorphisms: Cancer incidence and therapy. Oncogene 2006, 25, 1639–1648. [Google Scholar]
- Schumaker, L; Nikitakis, N; Goloubeva, O; Tan, M; Taylor, R; Cullen, KJ. Elevated expression of glutathione s-transferase pi and p53 confers poor prognosis in head and neck cancer patients treated with chemoradiotherapy but not radiotherapy alone. Clin. Cancer Res 2008, 14, 5877–5883. [Google Scholar]
- Ali-Osman, F; Akande, O; Antoun, G; Mao, JX; Buolamwini, J. Molecular cloning, characterization, and expression in escherichia coli of full-length cdnas of three human glutathione s-transferase pi gene variants. Evidence for differential catalytic activity of the encoded proteins. J. Biol. Chem 1997, 272, 10004–10012. [Google Scholar]
- Goto, S; Iida, T; Cho, S; Oka, M; Kohno, S; Kondo, T. Overexpression of glutathione s-transferase pi enhances the adduct formation of cisplatin with glutathione in human cancer cells. Free Radic. Res 1999, 31, 549–558. [Google Scholar]
- Stoehlmacher, J; Park, DJ; Zhang, W; Groshen, S; Tsao-Wei, DD; Yu, MC; Lenz, HJ. Association between glutathione s-transferase p1, t1, and m1 genetic polymorphism and survival of patients with metastatic colorectal cancer. J. Natl. Cancer Inst 2002, 94, 936–942. [Google Scholar]
- Maugard, CM; Charrier, J; Pitard, A; Campion, L; Akande, O; Pleasants, L; Ali-Osman, F. Genetic polymorphism at the glutathione s-transferase (gst) p1 locus is a breast cancer risk modifier. Int. J. Cancer 2001, 91, 334–339. [Google Scholar]
- Tew, KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res 1994, 54, 4313–4320. [Google Scholar]
- Miura, K; Suzuki, S; Tanita, J; Shinkawa, H; Satoh, K; Tsuchida, S. Correlated expression of glutathione s-transferase-pi and c-jun or other oncogene products in human squamous cell carcinomas of the head and neck: Relevance to relapse after radiation therapy. Jpn. J. Cancer Res 1997, 88, 143–151. [Google Scholar]
- Bongers, V; Snow, GB; de Vries, N; Cattan, AR; Hall, AG; van der Waal, I; Braakhuis, BJ. Second primary head and neck squamous cell carcinoma predicted by the glutathione s-transferase expression in healthy tissue in the direct vicinity of the first tumor. Lab. Invest 1995, 73, 503–510. [Google Scholar]
- Bentz, BG; Haines, GK, III; Lingen, MW; Pelzer, HJ; Hanson, DG; Radosevich, JA. Nitric oxide synthase type 3 is increased in squamous hyperplasia, dysplasia, and squamous cell carcinoma of the head and neck. Ann. Otol. Rhinol. Laryngol 1999, 108, 781–787. [Google Scholar]
- Bentz, BG; Haines, GK, III; Radosevich, JA. Glutathione s-transferase pi in squamous cell carcinoma of the head and neck. Laryngoscope 2000, 110, 1642–1647. [Google Scholar]
- Bentz, BG; Haines, GK, III; Lingen, MW; Pelzer, HJ; Hanson, DG; Radosevich, JA. Nitric oxide synthase type 3 is increased in squamous hyperplasia, dysplasia, and squamous cell carcinoma of the head and neck. Ann. Otol. Rhinol. Laryngol 1999, 108, 781–787. [Google Scholar]
- Bentz, BG; Haines, GK, III; Radosevich, JA. Glutathione s-transferase pi in squamous cell carcinoma of the head and neck. Laryngoscope 2000, 110, 1642–1647. [Google Scholar]
- Wei, L; Gravitt, PE; Song, H; Maldonado, AM; Ozbun, MA. Nitric oxide induces early viral transcription coincident with increased DNA damage and mutation rates in human papillomavirus-infected cells. Cancer Res 2009, 69, 4878–4884. [Google Scholar]
- Benencia, F; Gamba, G; Cavalieri, H; Courreges, MC; Benedetti, R; Villamil, SM; Massouh, EJ. Nitric oxide and hsv vaginal infection in balb/c mice. Virology 2003, 309, 75–84. [Google Scholar]
- Carratelli, CR; Rizzo, A; Paolillo, R; Catania, MR; Catalanotti, P; Rossano, F. Effect of nitric oxide on the growth of chlamydophila pneumoniae. Can. J. Microbiol 2005, 51, 941–947. [Google Scholar]
- Chang, K; Lubo, Z. Review article: Steroid hormones and uterine vascular adaptation to pregnancy. Reprod. Sci 2008, 15, 336–348. [Google Scholar]
- Hiraku, Y; Tabata, T; Ma, N; Murata, M; Ding, X; Kawanishi, S. Nitrative and oxidative DNA damage in cervical intraepithelial neoplasia associated with human papilloma virus infection. Cancer Sci 2007, 98, 964–972. [Google Scholar]
- Tavares-Murta, BM; de Resende, AD; Cunha, FQ; Murta, EF. Local profile of cytokines and nitric oxide in patients with bacterial vaginosis and cervical intraepithelial neoplasia. Eur. J. Obstet. Gynecol. Reprod. Biol 2008, 138, 93–99. [Google Scholar]
- Cerutti, PA. Prooxidant states and tumor promotion. Science 1985, 227, 375–381. [Google Scholar]
- Hussain, SP; Harris, CC. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar]
- Hussain, SP; Raja, K; Amstad, PA; Sawyer, M; Trudel, LJ; Wogan, GN; Hofseth, LJ; Shields, PG; Billiar, TR; Trautwein, C; et al. Increased p53 mutation load in nontumorous human liver of wilson disease and hemochromatosis: Oxyradical overload diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 12770–12775. [Google Scholar]
- Nair, J; Gansauge, F; Beger, H; Dolara, P; Winde, G; Bartsch, H. Increased etheno-DNA adducts in affected tissues of patients suffering from crohn's disease, ulcerative colitis, and chronic pancreatitis. Antioxid. Redox. Signal 2006, 8, 1003–1010. [Google Scholar]
- Cerutti, PA; Trump, BF. Inflammation and oxidative stress in carcinogenesis. Cancer Cells 1991, 3, 1–7. [Google Scholar]
- Shaulian, E; Karin, M. Ap-1 as a regulator of cell life and death. Nat. Cell Biol 2002, 4, E131–E136. [Google Scholar]
- Hofseth, LJ; Saito, S; Hussain, SP; Espey, MG; Miranda, KM; Araki, Y; Jhappan, C; Higashimoto, Y; He, P; Linke, SP; et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc. Natl. Acad. Sci. USA 2003, 100, 143–148. [Google Scholar]
- Ying, L; Marino, J; Hussain, SP; Khan, MA; You, S; Hofseth, AB; Trivers, GE; Dixon, DA; Harris, CC; Hofseth, LJ. Chronic inflammation promotes retinoblastoma protein hyperphosphorylation and e2f1 activation. Cancer Res 2005, 65, 9132–9136. [Google Scholar]
- Wink, DA; Hanbauer, I; Grisham, MB; Laval, F; Nims, RW; Laval, J; Cook, J; Pacelli, R; Liebmann, J; Krishna, M; et al. Chemical biology of nitric oxide: Regulation and protective and toxic mechanisms. Curr. Top Cell Regul 1996, 34, 159–187. [Google Scholar]
- Xu, W; Liu, L; Smith, GC; Charles, G. Nitric oxide upregulates expression of DNA-pkcs to protect cells from DNA-damaging anti-tumour agents. Nat. Cell Biol 2000, 2, 339–345. [Google Scholar]
- Jablonka, E; Lamb, MJ. The changing concept of epigenetics. Ann. NY Acad. Sci 2002, 981, 82–96. [Google Scholar]
- Peters, J. Overview of mammalian genome special issue on epigenetics. Mamm. Genome 2009, 20, 529–531. [Google Scholar]
- Waddington, CH. The epigenotype. Endeavour 1942, 18–20. [Google Scholar]
- Das, PM; Singal, R. DNA methylation and cancer. J. Clin. Oncol 2004, 22, 4632–4642. [Google Scholar]
- DeAngelis, JT; Farrington, WJ; Tollefsbol, TO. An overview of epigenetic assays. Mol. Biotechnol 2008, 38, 179–183. [Google Scholar]
- Jacob, S; Moley, KH. Gametes and embryo epigenetic reprogramming affect developmental outcome: Implication for assisted reproductive technologies. Pediatr. Res 2005, 58, 437–446. [Google Scholar]
- Tang, WY; Ho, SM. Epigenetic reprogramming and imprinting in origins of disease. Rev. Endocr. Metab. Disord 2007, 8, 173–182. [Google Scholar]
- Miller, CA; Sweatt, JD. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar]
- Matouk, CC; Marsden, PA. Epigenetic regulation of vascular endothelial gene expression. Circ. Res 2008, 102, 873–887. [Google Scholar]
- Prokhortchouk, E; Defossez, PA. The cell biology of DNA methylation in mammals. Biochim. Biophys. Acta 2008, 1783, 2167–2173. [Google Scholar]
- Horn, PJ; Peterson, CL. Molecular biology. Chromatin higher order folding--wrapping up transcription. Science 2002, 297, 1824–1827. [Google Scholar]
- Carthew, RW; Sontheimer, EJ. Origins and mechanisms of mirnas and sirnas. Cell 2009, 136, 642–655. [Google Scholar]
- Luger, K; Mader, AW; Richmond, RK; Sargent, DF; Richmond, TJ. Crystal structure of the nucleosome core particle at 2.8 a resolution. Nature 1997, 389, 251–260. [Google Scholar]
- Hansen, JC. Conformational dynamics of the chromatin fiber in solution: Determinants, mechanisms, and functions. Ann. Rev. Biophys. Biomol. Struct 2002, 31, 361–392. [Google Scholar]
- Jenuwein, T; Allis, CD. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar]
- Takai, D; Jones, PA. The cpg island searcher: A new www resource. In Silico Biol 2003, 3, 235–240. [Google Scholar]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet 2007, 8, 286–298. [Google Scholar]
- Jones, PA; Baylin, SB. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet 2002, 3, 415–428. [Google Scholar]
- Klose, RJ; Bird, AP. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci 2006, 31, 89–97. [Google Scholar]
- Takai, D; Jones, PA. Comprehensive analysis of cpg islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar]
- Catteau, A; Morris, JR. Brca1 methylation: A significant role in tumour development? Semin. Cancer Biol 2002, 12, 359–371. [Google Scholar]
- Egger, G; Liang, G; Aparicio, A; Jones, PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar]
- Leone, G; Teofili, L; Voso, MT; Lubbert, M. DNA methylation and demethylating drugs in myelodysplastic syndromes and secondary leukemias. Haematologica 2002, 87, 1324–1341. [Google Scholar]
- Tsou, JA; Hagen, JA; Carpenter, CL; Laird-Offringa, IA. DNA methylation analysis: A powerful new tool for lung cancer diagnosis. Oncogene 2002, 21, 5450–5461. [Google Scholar]
- Liu, J; Casaccia, P. Epigenetic regulation of oligodendrocyte identity. Trends Neurosci 2010, 33, 193–201. [Google Scholar]
- Chuang, JC; Jones, PA. Epigenetics and micrornas. Pediatr. Res 2007, 61, 24R–29R. [Google Scholar]
- Goel, A. Cpg island methylator phenotype in colrectal cancer: A current perspective. Curr Colorect Cancer Rep 2008, 77–83. [Google Scholar]
- Boland, R. Promoter methylation in the genesis of gastrointestinal cancer. Yonsei Med. J 2009, 50, 309–321. [Google Scholar]
- Vogelstein, B; Kinzler, KW. Cancer genes and the pathways they control. Nat. Med 2004, 10, 789–799. [Google Scholar]
- Bedford, MT; van Helden, PD. Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res 1987, 47, 5274–5276. [Google Scholar]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar]
- Kim, YI; Giuliano, A; Hatch, KD; Schneider, A; Nour, MA; Dallal, GE; Selhub, J; Mason, JB. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 1994, 74, 893–899. [Google Scholar]
- Lin, CH; Hsieh, SY; Sheen, IS; Lee, WC; Chen, TC; Shyu, WC; Liaw, YF. Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res 2001, 61, 4238–4243. [Google Scholar]
- Kawakami, K; Brabender, J; Lord, RV; Groshen, S; Greenwald, BD; Krasna, MJ; Yin, J; Fleisher, AS; Abraham, JM; Beer, DG; et al. Hypermethylated apc DNA in plasma and prognosis of patients with esophageal adenocarcinoma. J. Natl. Cancer Inst 2000, 92, 1805–1811. [Google Scholar]
- Harden, SV; Tokumaru, Y; Westra, WH; Goodman, S; Ahrendt, SA; Yang, SC; Sidransky, D. Gene promoter hypermethylation in tumors and lymph nodes of stage i lung cancer patients. Clin. Cancer Res 2003, 9, 1370–1375. [Google Scholar]
- Virmani, AK; Rathi, A; Sathyanarayana, UG; Padar, A; Huang, CX; Cunnigham, HT; Farinas, AJ; Milchgrub, S; Euhus, DM; Gilcrease, M; et al. Aberrant methylation of the adenomatous polyposis coli (apc) gene promoter 1a in breast and lung carcinomas. Clin. Cancer Res 2001, 7, 1998–2004. [Google Scholar]
- Chan, KY; Ozcelik, H; Cheung, AN; Ngan, HY; Khoo, US. Epigenetic factors controlling the brca1 and brca2 genes in sporadic ovarian cancer. Cancer Res 2002, 62, 4151–4156. [Google Scholar]
- Dobrovic, A; Simpfendorfer, D. Methylation of the brca1 gene in sporadic breast cancer. Cancer Res 1997, 57, 3347–3350. [Google Scholar]
- Sanchez-Cespedes, M; Esteller, M; Wu, L; Nawroz-Danish, H; Yoo, GH; Koch, WM; Jen, J; Herman, JG; Sidransky, D. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res 2000, 60, 892–895. [Google Scholar]
- Villuendas, R. Loss of p16/ink4a protein expression in non-hodgkin's lymphomas is a frequent finding associated with tumor progression. Am J Pathol 1998, 887–897. [Google Scholar]
- Graff, JR; Herman, JG; Lapidus, RG; Chopra, H; Xu, R; Jarrard, DF; Isaacs, WB; Pitha, PM; Davidson, NE; Baylin, SB. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995, 55, 5195–5199. [Google Scholar]
- Graff, JR; Greenberg, VE; Herman, JG; Westra, WH; Boghaert, ER; Ain, KB; Saji, M; Zeiger, MA; Zimmer, SG; Baylin, SB. Distinct patterns of e-cadherin cpg island methylation in papillary, follicular, hurthle's cell, and poorly differentiated human thyroid carcinoma. Cancer Res 1998, 58, 2063–2066. [Google Scholar]
- Waki, T; Tamura, G; Tsuchiya, T; Sato, K; Nishizuka, S; Motoyama, T. Promoter methylation status of e-cadherin, hmlh1, and p16 genes in nonneoplastic gastric epithelia. Am. J. Pathol 2002, 161, 399–403. [Google Scholar]
- Yang, X; Yan, L; Davidson, NE. DNA methylation in breast cancer. Endocr. Relat. Cancer 2001, 8, 115–127. [Google Scholar]
- Li, LC; Chui, R; Nakajima, K; Oh, BR; Au, HC; Dahiya, R. Frequent methylation of estrogen receptor in prostate cancer: Correlation with tumor progression. Cancer Res 2000, 60, 702–706. [Google Scholar]
- Lee, WH; Morton, RA; Epstein, JI; Brooks, JD; Campbell, PA; Bova, GS; Hsieh, WS; Isaacs, WB; Nelson, WG. Cytidine methylation of regulatory sequences near the pi-class glutathione s-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11733–11737. [Google Scholar]
- Veigl, ML; Kasturi, L; Olechnowicz, J; Ma, AH; Lutterbaugh, JD; Periyasamy, S; Li, GM; Drummond, J; Modrich, PL; Sedwick, WD; et al. Biallelic inactivation of hmlh1 by epigenetic gene silencing, a novel mechanism causing human msi cancers. Proc. Natl. Acad. Sci. USA 1998, 95, 8698–8702. [Google Scholar]
- Kondo, E. Not hmsh2 but hmlh1 is frequently silenced by hypermethylation in endometrial cancer but rarely silenced in pancreatic cancer with microsatellite instability. Int J Oncol 2000, 535–541. [Google Scholar]
- Strathdee, G; MacKean, MJ; Illand, M; Brown, R. A role for methylation of the hmlh1 promoter in loss of hmlh1 expression and drug resistance in ovarian cancer. Oncogene 1999, 18, 2335–2341. [Google Scholar]
- Harden, SV; Tokumaru, Y; Westra, WH; Goodman, S; Ahrendt, SA; Yang, SC; Sidransky, D. Gene promoter hypermethylation in tumors and lymph nodes of stage I lung cancer patients. Clin. Cancer Res 2003, 9, 1370–1375. [Google Scholar]
- Esteller, M; Garcia-Foncillas, J; Andion, E; Goodman, SN; Hidalgo, OF; Vanaclocha, V; Baylin, SB; Herman, JG. Inactivation of the DNA-repair gene mgmt and the clinical response of gliomas to alkylating agents. N. Engl. J. Med 2000, 343, 1350–1354. [Google Scholar]
- Melki, JR; Vincent, PC; Clark, SJ. Concurrent DNA hypermethylation of multiple genes in acute myeloid leukemia. Cancer Res 1999, 59, 3730–3740. [Google Scholar]
- Garcia, MJ; Martinez-Delgado, B; Cebrian, A; Martinez, A; Benitez, J; Rivas, C. Different incidence and pattern of p15ink4b and p16ink4a promoter region hypermethylation in hodgkin's and cd30-positive non-hodgkin's lymphomas. Am. J. Pathol 2002, 161, 1007–1013. [Google Scholar]
- Herman, JG; Jen, J; Merlo, A; Baylin, SB. Hypermethylation-associated inactivation indicates a tumor suppressor role for p15ink4b. Cancer Res 1996, 56, 722–727. [Google Scholar]
- Agathanggelou, A. Methylation associated inactivation of rassf1a from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 2001, 1509–1518. [Google Scholar]
- Morrissey, C; Martinez, A; Zatyka, M; Agathanggelou, A; Honorio, S; Astuti, D; Morgan, NV; Moch, H; Richards, FM; Kishida, T; et al. Epigenetic inactivation of the rassf1a 3p21.3 tumor suppressor gene in both clear cell and papillary renal cell carcinoma. Cancer Res 2001, 61, 7277–7281. [Google Scholar]
- Kwong, J; Lo, KW; To, KF; Teo, PM; Johnson, PJ; Huang, DP. Promoter hypermethylation of multiple genes in nasopharyngeal carcinoma. Clin. Cancer Res 2002, 8, 131–137. [Google Scholar]
- Stirzaker, C; Millar, DS; Paul, CL; Warnecke, PM; Harrison, J; Vincent, PC; Frommer, M; Clark, SJ. Extensive DNA methylation spanning the rb promoter in retinoblastoma tumors. Cancer Res 1997, 57, 2229–2237. [Google Scholar]
- Gonzalez-Gomez, P; Bello, MJ; Alonso, ME; Arjona, D; Lomas, J; de Campos, JM; Isla, A; Rey, JA. Cpg island methylation status and mutation analysis of the rb1 gene essential promoter region and protein-binding pocket domain in nervous system tumours. Br. J. Cancer 2003, 88, 109–114. [Google Scholar]
- Tannenbaum, SR; White, FM. Regulation and specificity of s-nitrosylation and denitrosylation. ACS Chem. Biol 2006, 1, 615–618. [Google Scholar]
- Sbaa, E; Frerart, F; Feron, O. The double regulation of endothelial nitric oxide synthase by caveolae and caveolin: A paradox solved through the study of angiogenesis. Trends Cardiovasc. Med 2005, 15, 157–162. [Google Scholar]
- Minchenkova, LE; Ivanov, VI. Influence of reductants upon optical characteristics of the DNA-Cu2+ complex. Biopolymers 1967, 5, 615–625. [Google Scholar]
- Robbins, E; Fant, J; Norton, W. Intracellular iron-binding macromolecules in hela cells. Proc. Natl. Acad. Sci. USA 1972, 69, 3708–3712. [Google Scholar]
- Bitny-Szlachto, S; Ochalska-Czepulis, M. Effects of disulphides and alpha-oxoglutarate on nuclear thiol formation and thiol content of chromatin in lysed rat spleen nuclei. Int. J. Biochem 1978, 9, 179–183. [Google Scholar]
- Vanin, AF; Ivanov, VI. Interaction of iron ions with oxygen or nitrogen monoxide in chromosomes triggers synchronous expression/suppression oscillations of compact gene groups (“Genomewide oscillation”): Hypothesis. Nitric Oxide 2008, 18, 147–152. [Google Scholar]
- Elzaouk, L; Laufs, S; Heerklotz, D; Leimbacher, W; Blau, N; Resibois, A; Thony, B. Nuclear localization of tetrahydrobiopterin biosynthetic enzymes. Biochim. Biophys. Acta 2004, 1670, 56–68. [Google Scholar]
- Calin, GA; Croce, CM. Microrna signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar]
- Nilsen, TW. Mechanisms of microrna-mediated gene regulation in animal cells. Trends Genet 2007, 23, 243–249. [Google Scholar]
- Farh, KK; Grimson, A; Jan, C; Lewis, BP; Johnston, WK; Lim, LP; Burge, CB; Bartel, DP. The widespread impact of mammalian micrornas on mrna repression and evolution. Science 2005, 310, 1817–1821. [Google Scholar]
- Lewis, BP; Shih, IH; Jones-Rhoades, MW; Bartel, DP; Burge, CB. Prediction of mammalian microrna targets. Cell 2003, 115, 787–798. [Google Scholar]
- Stark, A; Brennecke, J; Bushati, N; Russell, RB; Cohen, SM. Animal micrornas confer robustness to gene expression and have a significant impact on 3'utr evolution. Cell 2005, 123, 1133–1146. [Google Scholar]
- Dews, M; Homayouni, A; Yu, D; Murphy, D; Sevignani, C; Wentzel, E; Furth, EE; Lee, WM; Enders, GH; Mendell, JT; et al. Augmentation of tumor angiogenesis by a myc-activated microrna cluster. Nat. Genet 2006, 38, 1060–1065. [Google Scholar]
- O'Donnell, KA; Wentzel, EA; Zeller, KI; Dang, CV; Mendell, JT. C-myc-regulated micrornas modulate e2f1 expression. Nature 2005, 435, 839–843. [Google Scholar]
- Hayashita, Y; Osada, H; Tatematsu, Y; Yamada, H; Yanagisawa, K; Tomida, S; Yatabe, Y; Kawahara, K; Sekido, Y; Takahashi, T. A polycistronic microrna cluster, mir-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005, 65, 9628–9632. [Google Scholar]
- He, L; Thomson, JM; Hemann, MT; Hernando-Monge, E; Mu, D; Goodson, S; Powers, S; Cordon-Cardo, C; Lowe, SW; Hannon, GJ; et al. A microrna polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar]
- Lu, J; Getz, G; Miska, EA; Alvarez-Saavedra, E; Lamb, J; Peck, D; Sweet-Cordero, A; Ebert, BL; Mak, RH; Ferrando, AA; et al. Microrna expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar]
- Benetti, R; Gonzalo, S; Jaco, I; Munoz, P; Gonzalez, S; Schoeftner, S; Murchison, E; Andl, T; Chen, T; Klatt, P; et al. A mammalian microrna cluster controls DNA methylation and telomere recombination via rbl2-dependent regulation of DNA methyltransferases. Nat. Struct. Mol. Biol 2008, 15, 268–279. [Google Scholar]
- Sinkkonen, L; Hugenschmidt, T; Berninger, P; Gaidatzis, D; Mohn, F; Artus-Revel, CG; Zavolan, M; Svoboda, P; Filipowicz, W. Micrornas control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat. Struct. Mol. Biol 2008, 15, 259–267. [Google Scholar]
- Chan, JA; Krichevsky, AM; Kosik, KS. Microrna-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005, 65, 6029–6033. [Google Scholar]
- Chandra, RK; Bentz, BG; Haines, GK, III; Robinson, AM; Radosevich, JA. Expression of glutathione s-transferase pi in benign mucosa, barrett's metaplasia, and adenocarcinoma of the esophagus. Head Neck 2002, 24, 575–581. [Google Scholar]
- Voorhoeve, PM; le Sage, C; Schrier, M; Gillis, AJ; Stoop, H; Nagel, R; Liu, YP; van Duijse, J; Drost, J; Griekspoor, A; et al. A genetic screen implicates mirna-372 and mirna-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar]
- Eis, PS; Tam, W; Sun, L; Chadburn, A; Li, Z; Gomez, MF; Lund, E; Dahlberg, JE. Accumulation of mir-155 and bic rna in human b cell lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar]
- Iorio, MV; Ferracin, M; Liu, CG; Veronese, A; Spizzo, R; Sabbioni, S; Magri, E; Pedriali, M; Fabbri, M; Campiglio, M; et al. Microrna gene expression deregulation in human breast cancer. Cancer Res 2005, 65, 7065–7070. [Google Scholar]
- Tam, W; Dahlberg, JE. Mir-155/bic as an oncogenic microrna. Genes Chromos. Cancer 2006, 45, 211–212. [Google Scholar]
- Saito, Y; Liang, G; Egger, G; Friedman, JM; Chuang, JC; Coetzee, GA; Jones, PA. Specific activation of microrna-127 with downregulation of the proto-oncogene bcl6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar]
- Yoo, CB; Jones, PA. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov 2006, 5, 37–50. [Google Scholar]
- Weber, M; Baker, MB; Moore, JP; Searles, CD. Mir-21 is induced in endothelial cells by shear stress and modulates apoptosis and enos activity. Biochem. Biophys. Res. Commun 2010, 393, 643–648. [Google Scholar]
- Ji, R; Cheng, Y; Yue, J; Yang, J; Liu, X; Chen, H; Dean, DB; Zhang, C. Microrna expression signature and antisense-mediated depletion reveal an essential role of microrna in vascular neointimal lesion formation. Circ. Res 2007, 100, 1579–1588. [Google Scholar]
- Tatsuguchi, M; Seok, HY; Callis, TE; Thomson, JM; Chen, JF; Newman, M; Rojas, M; Hammond, SM; Wang, DZ. Expression of micrornas is dynamically regulated during cardiomyocyte hypertrophy. J. Mol. Cell Cardiol 2007, 42, 1137–1141. [Google Scholar]
- Wang, Y; Lee, CG. Microrna and cancer--focus on apoptosis. J. Cell Mol. Med 2009, 13, 12–23. [Google Scholar]
- Zeng, L; Carter, AD; Childs, SJ. Mir-145 directs intestinal maturation in zebrafish. Proc. Natl. Acad. Sci. USA 2009, 106, 17793–17798. [Google Scholar]
- Wang, X; Zhao, Q; Matta, R; Meng, X; Liu, X; Liu, CG; Nelin, LD; Liu, Y. Inducible nitric-oxide synthase expression is regulated by mitogen-activated protein kinase phosphatase-1. J. Biol. Chem 2009, 284, 27123–27134. [Google Scholar]
- Bui-Nguyen, TM; Pakala, SB; Sirigiri, DR; Martin, E; Murad, F; Kumar, R. Stimulation of inducible nitric oxide by hepatitis b virus transactivator protein hbx requires mta1 coregulator. J. Biol. Chem 2010, 285, 6980–6986. [Google Scholar]
- Bernstein, BE; Meissner, A; Lander, ES. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar]
- Chan, GC; Fish, JE; Mawji, IA; Leung, DD; Rachlis, AC; Marsden, PA. Epigenetic basis for the transcriptional hyporesponsiveness of the human inducible nitric oxide synthase gene in vascular endothelial cells. J. Immunol 2005, 175, 3846–3861. [Google Scholar]
- Goldberg, AD; Allis, CD; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar]
- Allis, CD; Berger, SL; Cote, J; Dent, S; Jenuwien, T; Kouzarides, T; Pillus, L; Reinberg, D; Shi, Y; Shiekhattar, R; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar]
- Yang, XJ; Seto, E. The rpd3/hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol 2008, 9, 206–218. [Google Scholar]
- Smith, BC; Hallows, WC; Denu, JM. Mechanisms and molecular probes of sirtuins. Chem. Biol 2008, 15, 1002–1013. [Google Scholar]
- Wang, GG; Allis, CD; Chi, P. Chromatin remodeling and cancer, part I: Covalent histone modifications. Trends Mol. Med 2007, 13, 363–372. [Google Scholar]
- Sengupta, N; Seto, E. Regulation of histone deacetylase activities. J. Cell Biochem 2004, 93, 57–67. [Google Scholar]
- Stamler, JS; Singel, DJ; Loscalzo, J. Biochemistry of nitric oxide and its redox-activated forms. Science 1992, 258, 1898–1902. [Google Scholar]
- Yang, XJ; Gregoire, S. Class ii histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell Biol 2005, 25, 2873–2884. [Google Scholar]
- Lin, J; Handschin, C; Spiegelman, BM. Metabolic control through the pgc-1 family of transcription coactivators. Cell Metab 2005, 1, 361–370. [Google Scholar]
- Fraga, MF; Esteller, M. DNA methylation: A profile of methods and applications. BioTechniques 2002, 33. [Google Scholar]
- Singal, R; Ginder, GD. DNA methylation. Blood 1999, 93, 4059–4070. [Google Scholar]
- Nestor, C. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. BioTechniques 2010, 48, 317–319. [Google Scholar]
- Chan, Y; Fish, JE; D'Abreo, C; Lin, S; Robb, GB; Teichert, AM; Karantzoulis-Fegaras, F; Keightley, A; Steer, BM; Marsden, PA. The cell-specific expression of endothelial nitric-oxide synthase: A role for DNA methylation. J. Biol. Chem 2004, 279, 35087–35100. [Google Scholar]
- Guillot, PV; Liu, L; Kuivenhoven, JA; Guan, J; Rosenberg, RD; Aird, WC. Targeting of human enos promoter to the hprt locus of mice leads to tissue-restricted transgene expression. Physiol. Genomics 2000, 2, 77–83. [Google Scholar]
- Teichert, AM; Miller, TL; Tai, SC; Wang, Y; Bei, X; Robb, GB; Phillips, MJ; Marsden, PA. In vivo expression profile of an endothelial nitric oxide synthase promoter-reporter transgene. Am. J. Physiol. Heart Circ. Physiol 2000, 278, H1352–1361. [Google Scholar]
- Bentz, BG; Hammer, ND; Radosevich, JA; Haines, GK, III. Nitrosative stress induces DNA strand breaks but not caspase mediated apoptosis in a lung cancer cell line. J. Carcinog 2004, 3, 16. [Google Scholar]
- Radosevich, JA; Elseth, KM; Vesper, BJ; Tarjan, G; Haines, GK, III. Long-term adaptation of lung tumor cell lines with increasing concentrations of nitric oxide donor. Open Lung Cancer J 2009, 2, 35–44. [Google Scholar]
- Vesper, BJ; Elseth, KM; Tarjan, G; Haines, GK, III; Radosevich, JA. Long-term adaptation of breast tumor cell lines to high concentrations of nitric oxide. Tumor. Biol 2010, 31, 267–275. [Google Scholar]
- Ridnour, LA; Thomas, DD; Donzelli, S; Espey, MG; Roberts, DD; Wink, DA; Isenberg, JS. The biphasic nature of nitric oxide responses in tumor biology. Antioxid. Redox. Signal 2006, 8, 1329–1337. [Google Scholar]
- Wink, DA; Mitchell, JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med 1998, 25, 434–456. [Google Scholar]
- Schetter, AJ; Heegaard, NH; Harris, CC. Inflammation and cancer: Interweaving microrna, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar]






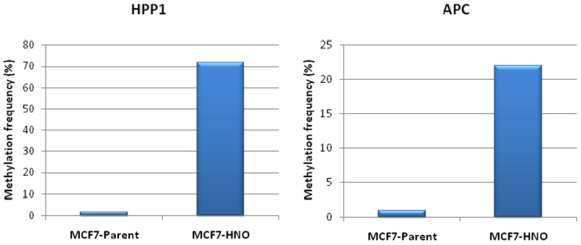
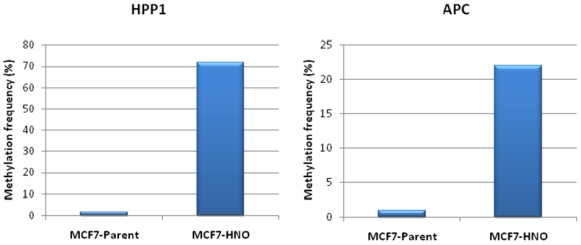{kind=link}
{kind=link}
{kind=link}
{kind=link}
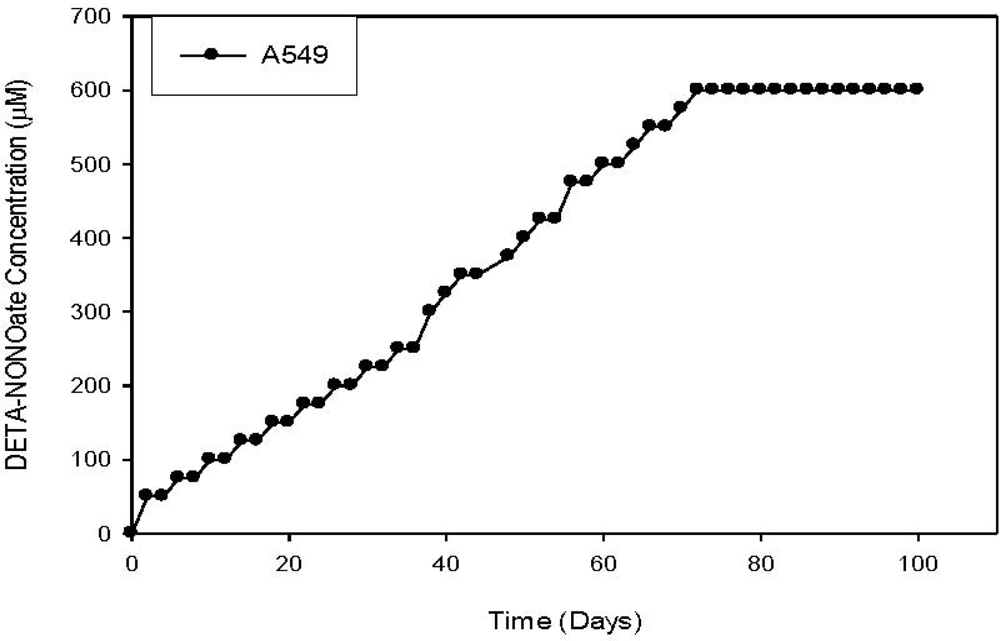{kind=link}
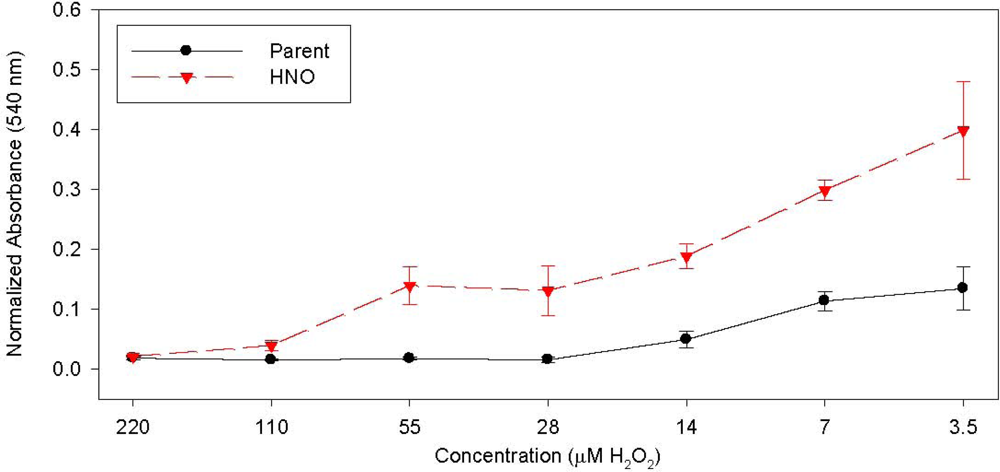{kind=link}
{kind=link}
| Patient | Age/Sex | Tumor Location | Tumor Stage | Surgery Performed | Previous Treatment | GST-pi Intensity | GST-pi Pattern |
|---|---|---|---|---|---|---|---|
| 1 | 74/M | Larynx | T3N0M0 (recurrent) | Total laryngectomy | Chemo/XRT | 3+ | diffuse |
| 2 | 79/F | Pyriform sinus | T3N0M0 | Laryngopharyngectomy | None | 1+ | focal |
| 3 | 73/M | Subglottis | T2N0M0 | Total laryngectomy | XRT | 3+ | diffuse |
| 4 | 75/F | Glottis | T4N0M0 | Laryngopharyngectomy | None | 1+ | diffuse |
| 5 | 73/M | Supraglottis | T2N0M0 (recurrent) | Supraglottic laryngectomy | XRT | 4+ | diffuse |
| 6 | 63/M | Supraglottis | T4N2M0 (recurrent) | Completion laryngectomy | Supraglottic laryngectomy, Chemo/XRT | 2+ | diffuse |
| 7 | 61/M | Supraglottis | T2N2M0 (recurrent) | Completion laryngectomy | Supraglottic laryngectomy, Chemo/XRT | 4+ | diffuse |
| 8 | 51/M | Supraglottis | T4N0M0 (recurrent) | Laryngopharyngectomy | Chemo/XRT | 3+ | focal |
| 9 | 77/M | Larynx | Recurrent | Total laryngectomy | Chemo/XRT | 3+ | diffuse |
| 10 | 81/M | Larynx | T3N0M0 (recurrent) | Completion laryngectomy | Supraglottic laryngectomy | 2+ | focal |
| Patient | Age | Stage | Grade | Treatment | Recurrence/Persistence | DFS (mos.) | iNOS Intensity | eNOS Intensity | GST-pi Intensity |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 39 | IIB | 2 | C-R | N | 32.5 | 3+ | 0 | 1+ |
| 2 | 48 | IV | 2 | N/A | N/A | N/A | 2+ | 0 | 1+ |
| 3 | 65 | IIIB | 3 | C-R | Y | 12.5 | 3+ | 0 | 0 |
| 4 | 41 | IIIB | 2 | C-R | Y | 4 | 2+ | 0 | 1+ |
| 5 | 39 | IIIB | 3 | C-R | N/A | N/A | 2+ | 1+ | 2+ |
| 6 | 50 | IB2 | 3 | C-R | N | 37 | 3+ | 2+ | 2+ |
| 7 | 38 | IB1 | 2 | S | N | 39 | 3+ | 1+ | 1+ |
| 8 | 49 | IB2 | 2 | C-R | N | 38 | 3+ | 1+ | 2+ |
| 9 | 63 | IIIB | 2 | C-R | Y | 9 | 1+ | 0 | 1+ |
| 10 | 29 | IIB | 2 | R | N/A | N/A | 2+ | 0 | 1+ |
| 11 | 49 | IIIB | 2 | C-R | Y | 8 | 2+ | 2+ | 1+ |
| 12 | 49 | IIB | 3 | C-R | N/A | N/A | 2+ | 0 | 2+ |
| 13 | 61 | IIIB | 3 | C-R | N/A | N/A | 2+ | 1+ | 2+ |
| 14 | 63 | IIB | 2 | C-R | N | 40 | 3+ | 2+ | 2+ |
| 15 | 44 | IIB | 2 | C-R | N | 42 | 3+ | 1+ | 2+ |
| 16 | 44 | N/A | 2 | N/A | N/A | N/A | 2+ | 1+ | 1+ |
| 17 | 52 | IVA | 3 | N/A | N/A | N/A | 3+ | 1+ | 2+ |
| 18 | 39 | IIB | 2 | C-R | N | 39 | 2+ | 1+ | 2+ |
| 19 | 51 | IIIB | 2 | C-R | N/A | N/A | 2+ | 1+ | 1+ |
| 20 | 37 | IB1 | 2 | S-R | N | 35 | 2+ | 0 | 1+ |
| 21 | 54 | IB2 | 3 | C-R | N | 50 | 2+ | 1+ | 1+ |
| 22 | 49 | IIA | 3 | R | N | 49 | 2+ | 1+ | 1+ |
| 23 | 48 | IIB | 2 | C-R | N | 48 | 2+ | 0 | 2+ |
| Gene | Role/Function | Tumor Type/Location | Impact | Reference(s) |
|---|---|---|---|---|
| APC | Inhibitor of β-catenin | Aerodigestive tract, lung, breast | Activation β –catenin route | [90,107–109] |
| AR | Androgen receptor | Prostate | Hormone insensitivity | [90] |
| BRCA1 | DNA repair, transcription | Breast, ovarian | Double strand breaks | [90,110,111] |
| CDH1 | E cadherin, cell adhesion | Breast, stomach, Leukemia | Dissemination | [90] |
| CDH13 | H cadherin, cell division | Breast, lung | Dissemination | [90] |
| CDKN2A/p16 | Cyclin-dependent kinase inhibitor | Head, neck, gastrointestinal tract, lung, NHL | Entrance in cell cycle | [78,90,108,112,113] |
| COX2 | Cycloxyenase-2 | Colon, stomach | Anti-inflammatory resistance1 | [90] |
| CPBP1 | Retinol-binding protein | Colon, stomach, lymphoma | Vitamin insensitivity | [90] |
| DAPK1 | Pro-apoptotic | Lymphoma, lung, colon | Resistance to apoptosis | [90,108] |
| DKK1 | Extracellular Wnt inhibitor | Colon | Activation Wnt signaling | [90] |
| DNMT1 | DNA disruption | Various | Over-expression | [90] |
| DNMT3b | DNA disruption | Various | Over-expression | [90] |
| E-cadherin | Increasing proliferation, invasion and/or metastasis | Breast, Thyroid, Gastric | [114–116] | |
| ER | Oestrogen receptor | Breast, prostate | Hormone insensitivity | [117,118] |
| EXT1 | Heparan intermediate filament | Leukemia, skin | Cellular detachment | [90] |
| FAT | Cadherin, tumor suppressor | Colon | Dissemination | [90] |
| GATA4 | Transcription factor | Colon, stomach | Silencing of target genes | [90] |
| GATA5 | Transcription factor | Colon, stomach | Silencing of target genes | [90] |
| GSTP1 | Conjugation to glutathione | Prostate, breast, kidney | Adduct accumulation | [90,108,119] |
| HIC1 | Transcription factor | Various forms | Currently unknown | [90] |
| HOXA9 | Homeobox protein | Neuroblastoma | Currently unknown | [90] |
| hMLH1 | Defective DNA mismatch repair, gene mutations | Colon, Renal, Gastric, Endometrim, Ovarian | [116,120–122] | |
| ID4 | Transcription factor | Leukemia, stomach | Currently unknown | [90] |
| IGFBP3 | Growth factor binding protein | Lung, skin | Resistance to apoptosis | [90] |
| Lamin A/C | Nuclear intermediate filament | Lymphoma, leukemia | Currently unknown | [90] |
| LKB1/STK11 | Serine-theronine kinase | Colon, breast, lung | Currently unknown | [90] |
| MBD1 | Rare mutations | Various | Over-expression | [90] |
| MBD2 | Rare mutations | Various | Over-expression | [90] |
| MBD3 | Rare mutations | Various | Over-expression | [90] |
| MBD4 | Rare mutations | Various | Over-expression | [90] |
| MeCP2 | Rare mutations | Various | Over-expression | [90] |
| MGMT | DNA repair of 06-alkyl-guanine, p53 | Lung, brain, various | Mutations, chemosensitivity | [90,123,124] |
| MLH1 | DNA mismatch repair | Colon, endometrium, stomach, ovarian | Frameshift mutations, gene mutations | [90] |
| NORE1A | Ras effector homologue | Lung | Currently unknown | [90] |
| p14ARF | MDM2 inhibitor | Colon, stomach. kidney | Degradation of p53 | [90] |
| p15 | Leukemia, Lymphoma | Entrance in cell cycle | [125–127] | |
| p15INK4b | Cyclin-dependent kinase inhibitor | Leukemia, lymphoma, lung, SCC | Entrance in cell cycle | [90] |
| p16INK4a | Cyclin-dependent kinase inhibitor | Various | Entrance in cell cycle | [90,108] |
| p73 | P53 homologue | Lymphoma | Currently unknown | [90] |
| PR | Progestrogen receptor | Breast | Hormone insensitivity | [90] |
| PRLR | Prolactin receptor | Breast | Hormone insensitivity | [90] |
| RARβ2 | Retinoic acid receptor –β2 | Colon, lung, head and neck | Vitamin insensitivity | [90] |
| RASSF1A | Ras effector homologue | Lung, breast, ovarian, kidney, nasopharyngeal | Currently unknown | [128–130] |
| Rb | Cell-cycle inhibitor | Retinoblastoma, oligodenodroglioma | Entrance to cell | [90,131,132] |
| RIZ1 | Histone/protein methyltransferase | Breast, liver | Abnormal gene expression | [90] |
| SFRP1 | Secreted frizzled-related protein 1 | Colon | Activation Wnt signaling | [90] |
| SLC5A8 | Sodium transporter | Glioma, colon | Currently unknown | [90] |
| SOC1 | Inhibitor of JAK-STAT pathway | Liver, mieloma | JAK2 activation | [90] |
| SOC3 | Inhibitor of JAK-STAT pathway | Lung | JAK2 activation | [90] |
| SRBC | BRCA1-binding protein | Breast, lung | Currently unknown | [90] |
| SYK | Tyrosine kinase | Breast | Currently unknown | [90] |
| THBS1 | Thrombospondin-1, anti-angiogenic | Giloma | Neo-vascularization | [90] |
| TMS1 | Pro-apoptotic | Breast | Resistance to apoptosis | [90] |
| TPEF/HPP1 | Transmembrane protein | Colon, bladder | Currently unknown | [90] |
| TSHR | Thyroid-stimulating hormone receptor | Thyroid | Hormone insensitivity | [90] |
| VHL | Ubiquitin ligase component | Kidney, haemangioblastoma | Loss of hypoxic response | [90,129] |
| WIF1 | Wnt inhibitor factor | Colon, lung | Activation Wnt signaling | [90] |
| WRN | DNA repair | Colon, stomach, sarcoma | DNA breakage, chemosensitivity | [90] |
| Substrate | Modification | Effect on Nucleosome/Chromatin | Transcription |
|---|---|---|---|
| AP-1 | S-N | Indirect | - |
| AtMYB2 | S-N | Indirect | - |
| Class II HDACs | Dephosphorylation | Indirect | _ |
| c-Myb | S-N | Indirect | - |
| GR | T-N | Indirect | + |
| HDAC2 | S-N, T-N | Indirect | + |
| HIF-1α | S-N | Indirect | + |
| Histones | T-N | Direct | ? |
| ikBα | T-N | Indirect | + |
| NF-kB | S-N, T-N | Indirect | - |
| Notch | T-N | Indirect | - |
| Nuclear receptors | S-N | Indirect | - |
| OxyR and SoxR | S-N | Indirect | + |
| P53 | T-N | Indirect | - |
| PPARγ | T-N | Indirect | - |
| β –catenin | T-N | Indirect | - |
| Gene | Tumor Type/Location | Impact |
|---|---|---|
| CBP1 | Colon, stomach, endometrium, lung, leukemia | Mutations, translocations, deletions |
| EZH23 | Various types | Gene amplification, over-expression |
| GASC14 | Squamous cell carcinoma | Gene amplification |
| HDAC12 | Various types | Aberrant expression |
| HDAC22 | Various types | Aberrant expression, mutations in MSI+ |
| MLL13 | Haematological malignancies | Translocation |
| MLL23 | Glioma, pancreas | Gene amplification |
| MLL33 | Leukemia | Deletion |
| MORF1 | Haematological malignancies, leiomyomata | Translocations |
| MOZ1 | Haematological malignancies | Translocations |
| NSD13 | Leukemia | Translocation |
| p3001 | Colon, stomach, endometrium | Mutations in MSI+ |
| pCAF1 | Colon | Rare mutations |
| RIZ13 | Various types | CpG-island hypermethylation |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Paradise, W.A.; Vesper, B.J.; Goel, A.; Waltonen, J.D.; Altman, K.W.; Haines, G.K., III; Radosevich, J.A. Nitric Oxide: Perspectives and Emerging Studies of a Well Known Cytotoxin. Int. J. Mol. Sci. 2010, 11, 2715-2745. https://doi.org/10.3390/ijms11072715
Paradise WA, Vesper BJ, Goel A, Waltonen JD, Altman KW, Haines GK III, Radosevich JA. Nitric Oxide: Perspectives and Emerging Studies of a Well Known Cytotoxin. International Journal of Molecular Sciences. 2010; 11(7):2715-2745. https://doi.org/10.3390/ijms11072715
Chicago/Turabian StyleParadise, William A., Benjamin J. Vesper, Ajay Goel, Joshua D. Waltonen, Kenneth W. Altman, G. Kenneth Haines, III, and James A. Radosevich. 2010. "Nitric Oxide: Perspectives and Emerging Studies of a Well Known Cytotoxin" International Journal of Molecular Sciences 11, no. 7: 2715-2745. https://doi.org/10.3390/ijms11072715
APA StyleParadise, W. A., Vesper, B. J., Goel, A., Waltonen, J. D., Altman, K. W., Haines, G. K., III, & Radosevich, J. A. (2010). Nitric Oxide: Perspectives and Emerging Studies of a Well Known Cytotoxin. International Journal of Molecular Sciences, 11(7), 2715-2745. https://doi.org/10.3390/ijms11072715