The “Autothixotropic” Phenomenon of Water and its Role in Proton Transfer

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discussion
2.1. Water as Liquid and Solvent
2.1.1. Hydrogen Bonds
2.1.2. Influence of Ions
2.1.3. Influence of Hydrophilic Surfaces
2.2. Time Related Changes in Aqueous Solutions
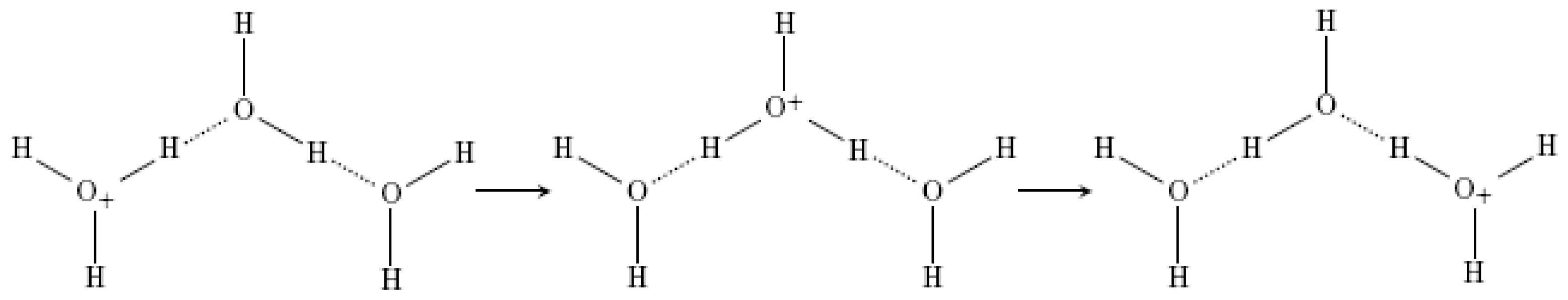
2.3. The Possible Role of the “Autothixotropic” Properties of Water on Living Beings and Proton Transfer
3. Conclusions
Acknowledgments
References
- Fisenko, A.I.; Malomuzh, N.P. The role of the H-bond network in the creation of the life-giving properties of water. Chem. Phys 2008, 345, 164–172. [Google Scholar]
- Henry, M. The state of water in living systems: from the liquid to the jellyfish. Cell Mol. Biol 2005, 51, 677–702. [Google Scholar]
- Henry, M. Water: Facts without Myths. Water 2009, 1, 3–4. [Google Scholar]
- Bizzarri, A.R.; Cannistraro, S. Molecular dynamics of water at the protein-solvent interface. J. Phys. Chem. B 2002, 106, 6617–6633. [Google Scholar]
- Zheng, J.M.; Pollack, G.H. Long-range forces extending from polymer-gel surfaces. Phys. Rev. E Stat. Nonlinear Soft Matter Phys 2003, 68, 031408–031414. [Google Scholar]
- Zheng, J.-M.; Chin, W.-C.; Khijniak, E.; Khijniak, E., Jr; Pollack, G.H. Surfaces and interfacial water: evidence that hydrophilic surfaces have long-range impact. Adv. Colloid Interface Sci. 2006, 127, 19–27. [Google Scholar]
- Guckenberger, R.; Heim, M.; Cevc, G.; Knapp, H.F.; Wiegräbe, W.; Hillebrand, A. Scanning tunneling microscopy of insulators and biological specimens based on lateral conductivity of ultrathin water films. Science 1994, 266, 1538–1540. [Google Scholar]
- Sasaki, N. Dielectric properties of slightly hydrated collagen: time-water content superposition analysis. Biopolymers 1984, 23, 1725–1734. [Google Scholar]
- Verdel, N.; Jerman, I.; Krašovec, R.; Bukovec, P. Influence of Water Ordering near Hydrophilic Surfaces on Conductivity and its Biological Importance. Proceedings of the Symposium Electrodynamic Activity of Living Cells, Prague, Czech Republic, 1–3 July 2011; Institute of Photonics and Electronics: Prague, Czech Republic, 2011; p. 24. [Google Scholar]
- Verdel, N.; Jerman, I.; Krašovec, R.; Bukovec, P.; Zupančič, M. Effect of ions and hydrophilic surfaces on the physic-chemical properties of water. J. Solut. Chem. 2011. submitted for publication. [Google Scholar]
- Elia, V.; Napoli, E.; Niccoli, M.; Marchettini, N.; Tiezzi, E. New physico-chemical properties of extremely diluted solutions. A conductivity study at 25°C in relation to ageing. J. Solut. Chem 2008, 37, 85–96. [Google Scholar]
- Elia, V.; Napoli, E.; Niccoli, M.; Nonatelli, L.; Ramaglia, A.; Ventimiglia, E. New physic-chemical properties of extremely diluted aqueous solutions. A calorimetric and conductivity study at 25 °C. J. Therm. Anal. Calorim 2004, 78, 331–342. [Google Scholar]
- Elia, V.; Marchese, M.; Montanino, M.; Napoli, E.; Niccoli, M.; Nonatelli, L.; Ramaglia, A. Hydrohysteretic phenomena of extremely diluted solutions induced by mechanical treatments: a calorimetric and conductometric study at 25 °C. J. Solut. Chem 2005, 34, 947–960. [Google Scholar]
- Elia, V.; Elia, L.; Montanino, M.; Napoli, E.; Niccoli, M.; Nonatelli, L. Conductometric studies of the serially diluted and agitated solutions on an anomalous effect that depends on the dilution process. J. Mol. Liq 2007, 135, 158–165. [Google Scholar]
- Elia, V.; Napoli, E.; Niccoli, M. A molecular model of interaction between extremely diluted solutions and NaOH solutions used as titrant. Conductometric and pHmetric titrations. J. Mol. Liq 2009, 148, 45–50. [Google Scholar]
- Elia, V.; Napoli, E.; Niccoli, M. Thermodynamic parameters for the binding process of the OH− ion with the dissipative structures. Calorimetric and conductometric titrations. J. Therm. Anal. Calorim 2010, 102, 1111–1118. [Google Scholar]
- Han, J.; Zhou, X.; Liu, H. Ab initio simulation on the mechanism of proton transport in water. J. Power Sources 2006, 161, 1420–1427. [Google Scholar]
- Xantheas, S.S. Computational chemistry: Dances with hydrogen cations. Nature 2009, 457, 673–674. [Google Scholar]
- Vybíral, B.; Voráček, P. Long term structural effects in water: autothixotropy of water and its hysteresis. Homeopathy 2007, 96, 183–188. [Google Scholar]
- Marcus, Y. The Properties of Solvents, 4th ed.; John Wiley & Sons: New York, NY, USA, 1998; pp. 1–97. [Google Scholar]
- Head-Gordon, T.; Hura, G. Water structure from scattering experiments and simulation. Chem. Rev 2002, 102, 2651–2670. [Google Scholar]
- Stillinger, F.H. Water revisited. Science 1980, 209, 451–457. [Google Scholar]
- Ball, P. Water as an active constituent in cell biology. Chem. Rev 2008, 108, 74–108. [Google Scholar]
- Xantheas, S.S. Cooperativity and hydrogen bonding network in water clusters. Chem. Phys 2000, 258, 225–231. [Google Scholar]
- Eaves, J.D.; Loparo, J.J.; Fecko, C.J.; Roberts, S.T.; Tokmakoff, A.; Geissler, P.L. Hydrogen bonds in liquid water are broken only fleetingly. Proc. Natl. Acad. Sci. USA 2005, 102, 13019–13022. [Google Scholar]
- Jenkins, H.D.B.; Marcus, Y. Viscosity B-coefficients of ions in solution. Chem. Rev 1995, 95, 2695–2724. [Google Scholar]
- Mancinelli, R.; Botti, A.; Bruni, F.; Ricci, M.A.; Soper, A.K. Perturbation of water structure due to monovalent ions in solutions. Phys. Chem. Chem. Phys 2007, 9, 2959–2967. [Google Scholar]
- Krekeler, C.; Delle Site, L. Solvation of positive ions in water: the dominant role of water-water interaction. J. Phys. Condens. Matter 2007, 19, 192101–192104. [Google Scholar]
- Omta, A.W.; Kropman, M.F.; Woutersen, S.; Bakker, H.J. Negligible effect of ions on the hydrogen-bond structure in liquid water. Science 2003, 301, 347–349. [Google Scholar]
- Turton, D.A.; Hunger, J.; Hefter, G.; Buchner, R.; Wynne, K. Glasslike behavior in aqueous electrolyte solutions. J. Chem. Phys 2008, 128, 161102–161105. [Google Scholar]
- Cheng, J.-X.; Pautot, S.; Weitz, D.A.; Xie, X.S. Ordering of water molecules between phospholipid bilayers visualized by CARS microscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 9826–9830. [Google Scholar]
- Jena, K.C.; Hore, D.K. Variation of ionic strength reveals the interfacial water structure at a charged mineral surface. J. Phys. Chem. C 2009, 113, 15364–15372. [Google Scholar]
- Rhykerd, C.L., Jr; Schoen, M.; Diestler, D.J.; Cushman, J.H. Classical fluids in micropores and near solid surfaces. Nature 1987, 330, 461–463. [Google Scholar]
- Zhao, Q.; Zheng, J.M.; Chai, B.H.; Pollack, G.H. Unexpected effect of light on colloidal crystal spacing. Langmuir 2008, 24, 1750–1755. [Google Scholar]
- Zhao, Q.; Ovchinnikova, K.; Chai, B.; Yoo, H.; Magula, J.; Pollack, G.H. Role of proton gradients in the mechanism of osmosis. J. Phys. Chem. B 2009, 113, 10708–10714. [Google Scholar]
- Ovchinnikova, K.; Pollack, G.H. Can water store charge? Langmuir 2009, 2, 542–547. [Google Scholar]
- Pollack, G.H.; Figueroa, X.; Zhao, Q. Molecules, water, and radiant energy: new clues for the origin of life. Int. J. Mol. Sci 2009, 10, 1419–1429. [Google Scholar]
- Chai, B.; Pollack, G.H. Solute-free interfacial zones in polar liquids. J. Phys. Chem. B 2010, 114, 5371–5375. [Google Scholar]
- Henniker, J. The depth of the surface zone of a liquid. Rev. Mod. Phys 1949, 21, 322–341. [Google Scholar]
- Voeikov, V.L.; del Giudice, E. Water respiration: the base of the living state. Water 2009, 1, 52–75. [Google Scholar]
- Vybíral, B. The Comprehensive Experimental Research on the Autothixotropy of Water. In Water and the Cell; Pollack, G.H., Cameron, I., Wheatley, D., Eds.; Springer: New York, NY, USA, 2006; pp. 299–314. [Google Scholar]
- Lobyshev, V.I.; Shikhlinskaya, R.E.; Ryzhikov, B.D. Experimental evidence for intrinsic luminescence of water. J. Mol. Liq 1999, 8, 73–81. [Google Scholar]
- Paracelsus. Paracelsus: Selected Writings; Jacobi, J., Ed.; Princeton University Press: Princeton, NJ, USA, 1979; p. 13. [Google Scholar]
- Gerstein, M.; Levitt, M. Simulating water and the molecules of life. Sci. Am 1998, 279, 101–105. [Google Scholar]
- Frauenfelder, H.; Chen, G.; Berendzen, J.; Fenimore, P.W.; Jansson, H.; McMahon, B.H.; Stroe, I.R.; Swenson, J.; Young, R.D. A unified model of protein dynamics. Proc. Natl. Acad. Sci. USA 2009, 106, 5129–5134. [Google Scholar]
- Johnson, M.E.; Malardie-Jugroot, C.; Murarka, R.K.; Head-Gordon, T. Hydration water dynamics near biological interfaces. J. Phys. Chem 2009, 113, 4082–4092. [Google Scholar]
- Rau, D.C.; Sidorova, N.Y. Diffusion of the restriction nuclease EcoRI along DNA. J. Mol. Biol 2010, 395, 408–416. [Google Scholar]
- Blainey, P.C.; Luo, G.; Kou, S.C.; Mangel, W.F.; Verdine, G.L.; Bagchi, B.; Xie, X.S. Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol 2010, 16, 1224–1229. [Google Scholar]
- Fuxreiter, M.; Mezel, M.; Simon, I.; Osman, R. Interfacial water as a “hydration fingerprint” in the noncognate complex of BamHI. Biophys. J 2005, 89, 903–911. [Google Scholar]
- Skelton, A.A.; Taining, L.; Walsh, T.R. Interplay of sequence, conformation, and binding at the Peptide-titania interface as mediated by water. ACS Appl. Mater. Interfaces 2009, 1, 1482–1491. [Google Scholar]
- Notman, R.; Walsh, T.R. Molecular dynamics studies of the interactions of water and amino acid analogues with quartz surfaces. Langmuir 2009, 25, 1638–1644. [Google Scholar]
- Samsonov, S.; Teyra, J.; Pisabarro, M.T. A molecular dynamics approach to study the importance of solvent in protein interactions. Proteins 2008, 73, 515–525. [Google Scholar]
- Naguib, N.; Ye, H.; Gogotsi, Y.; Yazicioglu, A.G.; Megaridis, C.M.; Yoshimura, M. Observation of water confined in nanometer channels of closed carbon nanotubes. Nano Lett 2004, 4, 2237–2243. [Google Scholar]
- de Grotthuss, C.J.T. Sur la décomposition de l‘eau et des corps qu’elle tient en dissolution à l’aide de l’électricité galvanique. Ann. Chim 1806, LVIII, 54–74. [Google Scholar]
- Agmon, N. The grotthuss mechanism. Chem. Phys. Lett 1995, 244, 456–462. [Google Scholar]
- Cukierman, S. Et tu, Grotthuss! and other unfinished stories. Biochim. Biophys. Acta 2006, 1757, 876–885. [Google Scholar]
- Eigen, M. Proton transfer, acid-base catalysis, and enzymatic hydrolysis. Part 1: Elementary processes. Angew. Chem. Int. Ed 1964, 3, 1–19. [Google Scholar]
- Zundel, G.; Metzer, H. Energiebänder der tunnelnden Überschuβ-Protonen in flüssigen Säuren. Eine IR-spektroskopische Untersuchung der Natur der Gruppierungen H5O2 +. Z. Phys. Chem 1968, 58, 225–245. [Google Scholar]
- Smondyrev, A.M.; Voth, G.A. Molecular dynamics simulation of proton transport near the surface of a phospholipid membrane. Biophys. J 2002, 82, 1460–1468. [Google Scholar]
- Agmon, N.; Goldberg, S.Y.; Huppert, D. Salt effect on transient proton transfer to solvent and microscopic proton mobility. J. Mol. Liq 1995, 64, 161–195. [Google Scholar]
- Markovitch, O.; Chen, H.; Izvekov, S.; Paesani, F.; Voth, G.A.; Agmon, N. Special pair dance and partner selection: elementary steps in proton transport in liquid water. J. Phys. Chem. B 2008, 112, 9456–9466. [Google Scholar]
- Mohammed, O.F.; Pines, D.; Pines, E.; Nibbering, E.T.J. Aqueous bimolecular proton transfer in acid-base neutralization. Chem. Phys 2007, 341, 240–257. [Google Scholar]
- Siwick, B.J.; Bakker, H.J. On the role of water in intermolecular proton-transfer reactions. J. Am. Chem. Soc 2007, 129, 13412–13420. [Google Scholar]
- Chen, H.; Voth, G.A.; Agmon, N. Kinetics of proton migration in liquid water. J. Phys. Chem. B 2010, 114, 333–339. [Google Scholar]
- Lapid, H.; Agmon, N.; Petersen, M.K.; Voth, G.A. A bond-order analysis of the mechanism for hydrated proton mobility in liquid water. J. Chem. Phys 2005, 122, 014506–014516. [Google Scholar]
- Tielrooij, K.J.; Timmer, R.L.A.; Bakker, H.J.; Bonn, M. Structure dynamics of the proton in liquid water probed with terahertz time-domain spectroscopy. Phys. Rev. Lett 2009, 102, 198303–198306. [Google Scholar]
- Ling, G.L. Life at the Cell and Bellow-Cell Level: The Hidden History of a Fundamental Revolution in Biology, 1st ed; Pacific Press: New York, NY, USA, 2001; pp. 1–373. [Google Scholar]
- Saenger, W. Circular hydrogen bonds. Nature 1979, 279, 343–344. [Google Scholar]
- Suzuki, T.; Sota, T. Circular hydrogen bond networks on the surface of β-ribofuranose in aqueous solution. J. Phys. Chem. B 2005, 109, 12603–12611. [Google Scholar]
- Seibold, S.A.; Mills, D.A.; Ferguson-Miller, S.; Cukier, R.I. Water chain formation and possible proton pumping routes in Rhodobacter sphaeroides cytochrome c oxidase: a molecular dynamics comparison of the wild type and R481K mutant. Biochemistry 2005, 44, 10475–10485. [Google Scholar]
- Ho, M.-W. The Rainbow and the Worm: The Physics of Organisms, 3rd ed.; World Scientific Publishing: Singapore, Singapore, 2008; pp. 256–260. [Google Scholar]


© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Verdel, N.; Jerman, I.; Bukovec, P. The “Autothixotropic” Phenomenon of Water and its Role in Proton Transfer. Int. J. Mol. Sci. 2011, 12, 7481-7494. https://doi.org/10.3390/ijms12117481
Verdel N, Jerman I, Bukovec P. The “Autothixotropic” Phenomenon of Water and its Role in Proton Transfer. International Journal of Molecular Sciences. 2011; 12(11):7481-7494. https://doi.org/10.3390/ijms12117481
Chicago/Turabian StyleVerdel, Nada, Igor Jerman, and Peter Bukovec. 2011. "The “Autothixotropic” Phenomenon of Water and its Role in Proton Transfer" International Journal of Molecular Sciences 12, no. 11: 7481-7494. https://doi.org/10.3390/ijms12117481