Oncolytic Activities of Host Defense Peptides

Abstract
:1. Introduction
1.1. Host Defense Peptides
1.2. Structure and Mechanism of Action
2. α-Helical Oncolytic Peptides
2.1. BMAP-27 & BMAP-28
2.2. Cecropin A and B
2.3. LL-37/hCAP-18
2.4. Magainins
2.5. Mellitin
3. B-Sheet Oncolytic Peptides
3.1. Defensins
3.2. Lactoferricin
3.3. Tachyplesin
4. Hybrid and Synthetic HDPs
4.1. Buforin II and Buforin IIb
4.2. Hunter-Killer Peptides
4.3. Polybia-MPI
4.4. Epinecidin-1
4.5. d,l-Amino Acids Host-Defence-like Peptides
5. Discussion
6. Conclusion
Acknowledgements
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol 2002, 3, 991–998. [Google Scholar]
- Wang, K.R.; Zhang, B.Z.; Zhang, W.; Yan, J.X.; Li, J.; Wang, R. Antitumor effects, cell selectivity and structure-activity relationship of a novel antimicrobial peptide polybia-MPI. Peptides 2008, 29, 963–968. [Google Scholar]
- Savarese, D.M.; Savy, G.; Vahdat, L.; Wischmeyer, P.E.; Corey, B. Prevention of chemotherapy and radiation toxicity with glutamine. Cancer Treat. Rev 2003, 29, 501–513. [Google Scholar]
- Perez-Tomas, R. Multidrug resistance: Retrospect and prospects in anti-cancer drug treatment. Curr. Med. Chem 2006, 13, 1859–1876. [Google Scholar]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci 2000, 11, 265–283. [Google Scholar]
- Papo, N.; Shai, Y. Host defense peptides as new weapons in cancer treatment. Cell. Mol. Life Sci 2005, 62, 784–790. [Google Scholar]
- Baker, M.A.; Maloy, W.L.; Zasloff, M.; Jacob, L.S. Anticancer efficacy of Magainin2 and analogue peptides. Cancer Res 1993, 53, 3052–3057. [Google Scholar]
- Boman, H.G.; Faye, I.; Gudmundsson, G.H.; Lee, J.Y.; Lidholm, D.A. Cell-free immunity in Cecropia. A model system for antibacterial proteins. Eur. J. Biochem 1991, 201, 23–31. [Google Scholar]
- Cruciani, R.A.; Barker, J.L.; Zasloff, M.; Chen, H.C.; Colamonici, O. Antibiotic magainins exert cytolytic activity against transformed cell lines through channel formation. Proc. Natl. Acad. Sci. USA 1991, 88, 3792–3796. [Google Scholar]
- Lichtenstein, A.; Ganz, T.; Selsted, M.E.; Lehrer, R.I. In vitro tumor cell cytolysis mediated by peptide defensins of human and rabbit granulocytes. Blood 1986, 68, 1407–1410. [Google Scholar]
- Lichtenstein, A.K.; Ganz, T.; Nguyen, T.M.; Selsted, M.E.; Lehrer, R.I. Mechanism of target cytolysis by peptide defensins. Target cell metabolic activities, possibly involving endocytosis, are crucial for expression of cytotoxicity. J. Immunol 1988, 140, 2686–2694. [Google Scholar]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Al-Benna, S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar]
- Steinstraesser, L.; Kraneburg, U.M.; Hirsch, T.; Kesting, M.; Steinau, H.U.; Jacobsen, F.; Al-Benna, S. Host defense peptides as effector molecules of the innate immune response: A sledgehammer for drug resistance? Int. J. Mol. Sci 2009, 10, 3951–3970. [Google Scholar]
- Andreu, D.; Rivas, L. Animal antimicrobial peptides: An overview. Biopolymers 1998, 47, 415–433. [Google Scholar]
- Hancock, R.E. Peptide antibiotics. Lancet 1997, 349, 418–422. [Google Scholar]
- Koczulla, A.R.; Bals, R. Antimicrobial peptides: Current status and therapeutic potential. Drugs 2003, 63, 389–406. [Google Scholar]
- van’t Hof, W.; Veerman, E.C.; Helmerhorst, E.J.; Amerongen, A.V. Antimicrobial peptides: Properties and applicability. Biol. Chem 2001, 382, 597–619. [Google Scholar]
- Iwasaki, T.; Ishibashi, J.; Tanaka, H.; Sato, M.; Asaoka, A.; Taylor, D.; Yamakawa, M. Selective cancer cell cytotoxicity of enantiomeric 9-mer peptides derived from beetle defensins depends on negatively charged phosphatidylserine on the cell surface. Peptides 2009, 30, 660–668. [Google Scholar]
- Matsuzaki, K.; Sugishita, K.; Ishibe, N.; Ueha, M.; Nakata, S.; Miyajima, K.; Epand, R.M. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry 1998, 37, 11856–11863. [Google Scholar]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides [Review]. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar]
- Bessalle, R.; Kapitkovsky, A.; Gorea, A.; Shalit, I.; Fridkin, M. All-d-magainin: Chirality, antimicrobial activity and proteolytic resistance. FEBS Lett 1990, 274, 151–155. [Google Scholar]
- Hetru, C.; Letellier, L.; Oren, Z.; Hoffmann, J.A.; Shai, Y. Androctonin, a hydrophilic disulphide-bridged non-haemolytic anti- microbial peptide: A plausible mode of action. Biochem. J 2000, 345, 653–664. [Google Scholar]
- Wade, D.; Boman, A.; Wahlin, B.; Drain, C.M.; Andreu, D.; Boman, H.G.; Merrifield, R.B. All-d amino acid-containing channel-forming antibiotic peptides. Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [Google Scholar]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.G.; Shai, Y. The Consequence of Sequence Alteration of an Amphipathic alpha -Helical Antimicrobial Peptide and Its Diastereomers. J. Biol. Chem 2002, 277, 33913–33921. [Google Scholar]
- Shai, Y.; Oren, Z. Diastereoisomers of cytolysins, a novel class of potent antibacterial peptides. J. Biol. Chem 1996, 271, 7305–7308. [Google Scholar]
- Chen, Y.; Xu, X.; Hong, S.; Chen, J.; Liu, N.; Underhill, C.B.; Creswell, K.; Zhang, L. RGD-Tachyplesin inhibits tumor growth. Cancer Res 2001, 61, 2434–2438. [Google Scholar]
- Ellerby, H.M.; Arap, W.; Ellerby, L.M.; Kain, R.; Andrusiak, R.; Rio, G.D.; Krajewski, S.; Lombardo, C.R.; Rao, R.; Ruoslahti, E.; et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med 1999, 5, 1032–1038. [Google Scholar]
- Papo, N.; Braunstein, A.; Eshhar, Z.; Shai, Y. Suppression of human prostate tumor growth in mice by a cytolytic d-, l-amino Acid Peptide: Membrane lysis, increased necrosis, and inhibition of prostate-specific antigen secretion. Cancer Res 2004, 64, 5779–5786. [Google Scholar]
- Papo, N.; Seger, D.; Makovitzki, A.; Kalchenko, V.; Eshhar, Z.; Degani, H.; Shai, Y. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res 2006, 66, 5371–5378. [Google Scholar]
- Papo, N.; Shahar, M.; Eisenbach, L.; Shai, Y. A novel lytic peptide composed of dl-amino acids selectively kills cancer cells in culture and in mice. J. Biol. Chem 2003, 278, 21018–21023. [Google Scholar]
- Papo, N.; Shai, Y. New lytic peptides based on the d,l-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry 2003, 42, 9346–9354. [Google Scholar]
- Chen, H.M.; Wang, W.; Smith, D.; Chan, S.C. Effects of the anti-bacterial peptide cecropin B and its analogs, cecropins B-1 and B-2, on liposomes, bacteria, and cancer cells. Biochim. Biophys. Acta 1997, 1336, 171–179. [Google Scholar]
- Eliassen, L.T.; Berge, G.; Sveinbjornsson, B.; Svendsen, J.S.; Vorland, L.H.; Rekdal, O. Evidence for a direct antitumor mechanism of action of bovine lactoferricin. Anticancer Res 2002, 22, 2703–2710. [Google Scholar]
- Johnstone, S.A.; Gelmon, K.; Mayer, L.D.; Hancock, R.E.; Bally, M.B. In vitro characterization of the anticancer activity of membrane-active cationic peptides. I. Peptide-mediated cytotoxicity and peptide-enhanced cytotoxic activity of doxorubicin against wild-type and p-glycoprotein over-expressing tumor cell lines. Anticancer Drug Des 2000, 15, 151–160. [Google Scholar]
- Ohsaki, Y.; Gazdar, A.F.; Chen, H.C.; Johnson, B.E. Antitumor activity of magainin analogues against human lung cancer cell lines. Cancer Res 1992, 52, 3534–3538. [Google Scholar]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res 1991, 51, 3062–3066. [Google Scholar]
- Yang, N.; Lejon, T.; Rekdal, O. Antitumour activity and specificity as a function of substitutions in the lipophilic sector of helical lactoferrin-derived peptide. J. Pept. Sci 2003, 9, 300–311. [Google Scholar]
- Yang, N.; Stensen, W.; Svendsen, J.S.; Rekdal, O. Enhanced antitumor activity and selectivity of lactoferrin-derived peptides. J. Pept. Res 2002, 60, 187–197. [Google Scholar]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1999, 1462, 11–28. [Google Scholar]
- Sitaram, N.; Nagaraj, R. Interaction of antimicrobial peptides with biological and model membranes: Structural and charge requirements for activity. Biochim. Biophys. Acta 1999, 1462, 29–54. [Google Scholar]
- Mai, J.C.; Mi, Z.; Kim, S.H.; Ng, B.; Robbins, P.D. A proapoptotic peptide for the treatment of solid tumors. Cancer Res 2001, 61, 7709–7712. [Google Scholar]
- Risso, A.; Braidot, E.; Sordano, M.C.; Vianello, A.; Macri, F.; Skerlavaj, B.; Zanetti, M.; Gennaro, R.; Bernardi, P. BMAP-28, an antibiotic peptide of innate immunity, induces cell death through opening of the mitochondrial permeability transition pore. Mol. Cell. Biol 2002, 22, 1926–1935. [Google Scholar]
- Takeshima, K.; Chikushi, A.; Lee, K.K.; Yonehara, S.; Matsuzaki, K. Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes. J. Biol. Chem 2003, 278, 1310–1315. [Google Scholar]
- Risso, A.; Zanetti, M.; Gennaro, R. Cytotoxicity and apoptosis mediated by two peptides of innate immunity. Cell. Immunol 1998, 189, 107–115. [Google Scholar]
- Skerlavaj, B.; Gennaro, R.; Bagella, L.; Merluzzi, L.; Risso, A.; Zanetti, M. Biological characterization of two novel cathelicidin-derived peptides and identification of structural requirements for their antimicrobial and cell lytic activities. J. Biol. Chem 1996, 271, 28375–28381. [Google Scholar]
- van Hofsten, P.; Faye, I.; Kockum, K.; Lee, J.Y.; Xanthopoulos, K.G.; Boman, I.A.; Boman, H.G.; Engstrom, A.; Andreu, D.; Merrifield, R.B. Molecular cloning, cDNA sequencing, and chemical synthesis of cecropin B from Hyalophora cecropia. Proc. Natl. Acad. Sci. USA 1985, 82, 2240–2243. [Google Scholar]
- Bechinger, B. Structure and functions of channel-forming peptides: Magainins, cecropins, melittin and alamethicin. J. Membr. Biol 1997, 156, 197–211. [Google Scholar]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med 2003, 254, 197–215. [Google Scholar]
- Hung, S.C.; Wang, W.; Chan, S.I.; Chen, H.M. Membrane lysis by the antibacterial peptides cecropins B1 and B3: A spin-label electron spin resonance study on phospholipid bilayers. Biophys. J 1999, 77, 3120–3133. [Google Scholar]
- Ye, J.S.; Zheng, X.J.; Leung, K.W.; Chen, H.M.; Sheu, F.S. Induction of transient ion channel-like pores in a cancer cell by antibiotic peptide. J. Biochem 2004, 136, 255–259. [Google Scholar]
- Hui, L.; Leung, K.; Chen, H.M. The combined effects of antibacterial peptide cecropin A and anti-cancer agents on leukemia cells. Anticancer Res 2002, 22, 2811–2816. [Google Scholar]
- Moore, A.J.; Devine, D.A.; Bibby, M.C. Preliminary experimental anticancer activity of cecropins. Pept. Res 1994, 7, 265–269. [Google Scholar]
- Wu, J.M.; Jan, P.S.; Yu, H.C.; Haung, H.Y.; Fang, H.J.; Chang, Y.I.; Cheng, J.W.; Chen, H.M. Structure and function of a custom anticancer peptide, CB1a. Peptides 2009, 30, 839–848. [Google Scholar]
- Shin, S.Y.; Lee, M.K.; Kim, K.L.; Hahm, K.S. Structure-antitumor and hemolytic activity relationships of synthetic peptides derived from cecropin A-magainin 2 and cecropin A-melittin hybrid peptides. J. Pept. Res 1997, 50, 279–285. [Google Scholar]
- Chan, S.C.; Hui, L.; Chen, H.M. Enhancement of the cytolytic effect of anti-bacterial cecropin by the microvilli of cancer cells. Anticancer Res 1998, 18, 4467–4474. [Google Scholar]
- Winder, D.; Gunzburg, W.H.; Erfle, V.; Salmons, B. Expression of antimicrobial peptides has an antitumour effect in human cells. Biochem. Biophys. Res. Commun 1998, 242, 608–612. [Google Scholar]
- Suttmann, H.; Retz, M.; Paulsen, F.; Harder, J.; Zwergel, U.; Kamradt, J.; Wullich, B.; Unteregger, G.; Stockle, M.; Lehmann, J. Antimicrobial peptides of the Cecropin-family show potent antitumor activity against bladder cancer cells. BMC Urol 2008, 8, 5:1–5:7. [Google Scholar]
- Ceron, J.M.; Contreras-Moreno, J.; Puertollano, E.; de Cienfuegos, G.A.; Puertollano, M.A.; de Pablo, M.A. The antimicrobial peptide cecropin A induces caspase-independent cell death in human promyelocytic leukemia cells. Peptides 2010, 31, 1494–1503. [Google Scholar]
- De, Y.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med 2000, 192, 1069–1074. [Google Scholar]
- Yang, D.; Chertov, O.; Oppenheim, J.J. The role of mammalian antimicrobial peptides and proteins in awakening of innate host defenses and adaptive immunity. Cell. Mol. Life Sci 2001, 58, 978–989. [Google Scholar]
- Steinstraesser, L.; Al-Benna, S.; Kesting, M.; Jacobsen, F. Bioengineered human skin: Working the bugs out. Mol. Ther 2009, 17, 405–408. [Google Scholar]
- Okumura, K.; Itoh, A.; Isogai, E.; Hirose, K.; Hosokawa, Y.; Abiko, Y.; Shibata, T.; Hirata, M.; Isogai, H. C-terminal domain of human CAP18 antimicrobial peptide induces apoptosis in oral squamous cell carcinoma SAS-H1 cells. Cancer Lett 2004, 212, 185–194. [Google Scholar]
- Li, X.; Li, Y.; Han, H.; Miller, D.W.; Wang, G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc 2006, 128, 5776–5785. [Google Scholar]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem 1998, 273, 3718–3724. [Google Scholar]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J 1999, 341, 501–513. [Google Scholar]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar]
- Maloy, W.L.; Kari, U.P. Structure-activity studies on magainins and other host defense peptides. Biopolymers 1995, 37, 105–122. [Google Scholar]
- Matsuzaki, K.; Sugishita, K.; Harada, M.; Fujii, N.; Miyajima, K. Interactions of an antimicrobial peptide, magainin 2, with outer and inner membranes of Gram-negative bacteria. Biochim. Biophys. Acta 1997, 1327, 119–130. [Google Scholar]
- Lehmann, J.; Retz, M.; Sidhu, S.S.; Suttmann, H.; Sell, M.; Paulsen, F.; Harder, J.; Unteregger, G.; Stockle, M. Antitumor activity of the antimicrobial peptide magainin II against bladder cancer cell lines. Eur. Urol 2006, 50, 141–147. [Google Scholar]
- Park, Y.; Lee, D.G.; Hahm, K.S. Antibiotic activity of Leu-Lys rich model peptides. Biotechnol. Lett 2003, 25, 1305–1310. [Google Scholar]
- Soballe, P.W.; Maloy, W.L.; Myrga, M.L.; Jacob, L.S.; Herlyn, M. Experimental local therapy of human melanoma with lytic magainin peptides. Int. J. Cancer 1995, 60, 280–284. [Google Scholar]
- Imura, Y.; Choda, N.; Matsuzaki, K. Magainin 2 in action: Distinct modes of membrane permeabilization in living bacterial and mammalian cells. Biophys. J 2008, 95, 5757–5765. [Google Scholar]
- Matsuzaki, K.; Harada, M.; Funakoshi, S.; Fujii, N.; Miyajima, K. Physicochemical determinants for the interactions of magainins 1 and 2 with acidic lipid bilayers. Biochim. Biophys. Acta 1991, 1063, 162–170. [Google Scholar]
- Peck-Miller, K.A.; Darveau, R.P.; Fell, H.P. Identification of serum components that inhibit the tumoricidal activity of amphiphilic alpha helical peptides. Cancer Chemother. Pharmacol 1993, 32, 109–115. [Google Scholar]
- Lincke, C.R.; van der Bliek, A.M.; Schuurhuis, G.J.; van der Velde-Koerts, T.; Smit, J.J.; Borst, P. Multidrug resistance phenotype of human BRO melanoma cells transfected with a wild-type human mdr1 complementary DNA. Cancer Res 1990, 50, 1779–1785. [Google Scholar]
- Endicott, J.A.; Ling, V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu. Rev. Biochem 1989, 58, 137–171. [Google Scholar]
- Sharma, S.V. Melittin resistance: A counterselection for ras transformation. Oncogene 1992, 7, 193–201. [Google Scholar]
- Hoover, D.M.; Boulegue, C.; Yang, D.; Oppenheim, J.J.; Tucker, K.; Lu, W.; Lubkowski, J. The structure of human macrophage inflammatory protein-3alpha/CCL20. Linking antimicrobial and CC chemokine receptor-6-binding activities with human beta-defensins. J. Biol. Chem 2002, 277, 37647–37654. [Google Scholar]
- Hoover, D.M.; Rajashankar, K.R.; Blumenthal, R.; Puri, A.; Oppenheim, J.J.; Chertov, O.; Lubkowski, J. The structure of human beta-defensin-2 shows evidence of higher order oligomerization. J. Biol. Chem 2000, 275, 32911–32918. [Google Scholar]
- Lehrer, R.I. Multispecific myeloid defensins. Curr. Opin. Hematol 2007, 14, 16–21. [Google Scholar]
- Pazgiera, M.; Hooverb, D.M.; Yangc, D.; Lud, W.; Lubkowskia, J. Human beta-defensins. Cell. Mol. Life Sci 2006, 63, 1294–1313. [Google Scholar]
- Bateman, A.; Singh, A.; Jothy, S.; Fraser, R.; Esch, F.; Solomon, S. The levels and biologic action of the human neutrophil granule peptide HP-1 in lung tumors. Peptides 1992, 13, 133–139. [Google Scholar]
- Chavakis, T.; Cines, D.B.; Rhee, J.S.; Liang, O.D.; Schubert, U.; Hammes, H.P.; Higazi, A.A.; Nawroth, P.P.; Preissner, K.T.; Bdeir, K. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): A link between inflammation and angiogenesis. FASEB J 2004, 18, 1306–1308. [Google Scholar]
- Economopoulou, M.; Bdeir, K.; Cines, D.B.; Fogt, F.; Bdeir, Y.; Lubkowski, J.; Lu, W.; Preissner, K.T.; Hammes, H.P.; Chavakis, T. Inhibition of pathologic retinal neovascularization by alpha-defensins. Blood 2005, 106, 3831–3838. [Google Scholar]
- Holterman, D.A.; Diaz, J.I.; Blackmore, P.F.; Davis, J.W.; Schellhammer, P.F.; Corica, A.; Semmes, O.J.; Vlahou, A. Overexpression of alpha-defensin is associated with bladder cancer invasiveness. Urol. Oncol 2006, 24, 97–108. [Google Scholar]
- Lundy, F.T.; Orr, D.F.; Gallagher, J.R.; Maxwell, P.; Shaw, C.; Napier, S.S.; Gerald Cowan, C.; Lamey, P.J.; Marley, J.J. Identification and overexpression of human neutrophil alpha-defensins (human neutrophil peptides 1, 2 and 3) in squamous cell carcinomas of the human tongue. Oral Oncol 2004, 40, 139–144. [Google Scholar]
- Muller, C.A.; Markovic-Lipkovski, J.; Klatt, T.; Gamper, J.; Schwarz, G.; Beck, H.; Deeg, M.; Kalbacher, H.; Widmann, S.; Wessels, J.T.; Becker, V.; Muller, G.A.; Flad, T. Human alpha-defensins HNPs-1, -2, and -3 in renal cell carcinoma: Influences on tumor cell proliferation. Am. J. Pathol 2002, 160, 1311–1324. [Google Scholar]
- Bullard, R.S.; Gibson, W.; Bose, S.K.; Belgrave, J.K.; Eaddy, A.C.; Wright, C.J.; Hazen-Martin, D.J.; Lage, J.M.; Keane, T.E.; Ganz, T.A.; et al. Functional analysis of the host defense peptide Human Beta Defensin-1: New insight into its potential role in cancer. Mol. Immunol 2008, 45, 839–848. [Google Scholar]
- Aarbiou, J.; Tjabringa, G.S.; Verhoosel, R.M.; Ninaber, D.K.; White, S.R.; Peltenburg, L.T.; Rabe, K.F.; Hiemstra, P.S. Mechanisms of cell death induced by the neutrophil antimicrobial peptides alpha-defensins and LL-37. Inflamm. Res 2006, 55, 119–127. [Google Scholar]
- Sheu, M.J.; Baldwin, W.W.; Brunson, K.W. Cytotoxicity of rabbit macrophage peptides MCP-1 and MCP-2 for mouse tumor cells. Antimicrob. Agents Chemother 1985, 28, 626–629. [Google Scholar]
- Kagan, B.L.; Selsted, M.E.; Ganz, T.; Lehrer, R.I. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc. Natl. Acad. Sci. USA 1990, 87, 210–214. [Google Scholar]
- Gera, J.F.; Lichtenstein, A. Human neutrophil peptide defensins induce single strand DNA breaks in target cells. Cell. Immunol 1991, 138, 108–120. [Google Scholar]
- Nishimura, M.; Abiko, Y.; Kurashige, Y.; Takeshima, M.; Yamazaki, M.; Kusano, K.; Saitoh, M.; Nakashima, K.; Inoue, T.; Kaku, T. Effect of defensin peptides on eukaryotic cells: Primary epithelial cells, fibroblasts and squamous cell carcinoma cell lines. J. Dermatol. Sci 2004, 36, 87–95. [Google Scholar]
- Wang, Y.S.; Li, D.; Shi, H.S.; Wen, Y.J.; Yang, L.; Xu, N.; Chen, X.C.; Chen, X.; Chen, P.; Li, J.; et al. Intratumoral expression of mature human neutrophil peptide-1 mediates antitumor immunity in mice. Clin. Cancer Res 2009, 15, 6901–6911. [Google Scholar]
- Biragyn, A. Defensins—Non-antibiotic use for vaccine development. Curr. Protein Pept. Sci 2005, 6, 53–60. [Google Scholar]
- Tani, K.; Murphy, W.J.; Chertov, O.; Salcedo, R.; Koh, C.Y.; Utsunomiya, I.; Funakoshi, S.; Asai, O.; Herrmann, S.H.; Wang, J.M.; et al. Defensins act as potent adjuvants that promote cellular and humoral immune responses in mice to a lymphoma idiotype and carrier antigens. Int. Immunol 2000, 12, 691–700. [Google Scholar]
- Hubert, P.; Herman, L.; Maillard, C.; Caberg, J.H.; Nikkels, A.; Pierard, G.; Foidart, J.M.; Noel, A.; Boniver, J.; Delvenne, P. Defensins induce the recruitment of dendritic cells in cervical human papillomavirus-associated (pre)neoplastic lesions formed in vitro and transplanted in vivo. FASEB J 2007, 21, 2765–2775. [Google Scholar]
- Conejo-Garcia, J.R.; Benencia, F.; Courreges, M.C.; Kang, E.; Mohamed-Hadley, A.; Buckanovich, R.J.; Holtz, D.O.; Jenkins, A.; Na, H.; Zhang, L.; et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat. Med 2004, 10, 950–958. [Google Scholar]
- Xu, N.; Wang, Y.S.; Pan, W.B.; Xiao, B.; Wen, Y.J.; Chen, X.C.; Chen, L.J.; Deng, H.X.; You, J.; Kan, B.; et al. Human alpha-defensin-1 inhibits growth of human lung adenocarcinoma xenograft in nude mice. Mol. Cancer Ther 2008, 7, 1588–1597. [Google Scholar]
- Bellamy, W.; Takase, M.; Yamauchi, K.; Wakabayashi, H.; Kawase, K.; Tomita, M. Identification of the bactericidal domain of lactoferrin. Biochim. Biophys. Acta 1992, 1121, 130–136. [Google Scholar]
- Cho, E.; Smith-Warner, S.A.; Spiegelman, D.; Beeson, W.L.; van den Brandt, P.A.; Colditz, G.A.; Folsom, A.R.; Fraser, G.E.; Freudenheim, J.L.; Giovannucci, E.; et al. Dairy foods, calcium, and colorectal cancer: A pooled analysis of 10 cohort studies. J. Natl. Cancer Inst 2004, 96, 1015–1022. [Google Scholar]
- Tsuda, H.; Sekine, K.; Nakamura, J.; Ushida, Y.; Kuhara, T.; Takasuka, N.; Kim, D.J.; Asamoto, M.; Baba-Toriyama, H.; Moore, M.A.; et al. Inhibition of azoxymethane initiated colon tumor and aberrant crypt foci development by bovine lactoferrin administration in F344 rats. Adv. Exp. Med. Biol 1998, 443, 273–284. [Google Scholar]
- Yoo, Y.C.; Watanabe, S.; Watanabe, R.; Hata, K.; Shimazaki, K.; Azuma, I. Bovine lactoferrin and lactoferricin, a peptide derived from bovine lactoferrin, inhibit tumor metastasis in mice. Jpn. J. Cancer Res 1997, 88, 184–190. [Google Scholar]
- Eliassen, L.T.; Berge, G.; Leknessund, A.; Wikman, M.; Lindin, I.; Lokke, C.; Ponthan, F.; Johnsen, J.I.; Sveinbjornsson, B.; Kogner, P.; et al. The antimicrobial peptide, lactoferricin B, is cytotoxic to neuroblastoma cells in vitro and inhibits xenograft growth in vivo. Int. J. Cancer 2006, 119, 493–500. [Google Scholar]
- Mader, J.S.; Richardson, A.; Salsman, J.; Top, D.; de Antueno, R.; Duncan, R.; Hoskin, D.W. Bovine lactoferricin causes apoptosis in Jurkat T-leukemia cells by sequential permeabilization of the cell membrane and targeting of mitochondria. Exp. Cell Res 2007, 313, 2634–2650. [Google Scholar]
- Perdigon, G.; de Moreno de LeBlanc, A.; Valdez, J.; Rachid, M. Role of yoghurt in the prevention of colon cancer. Eur. J. Clin. Nutr 2002, 56, S65–S68. [Google Scholar]
- Furlong, S.J.; Ridgway, N.D.; Hoskin, D.W. Modulation of ceramide metabolism in T-leukemia cell lines potentiates apoptosis induced by the cationic antimicrobial peptide bovine lactoferricin. Int. J. Oncol 2008, 32, 537–544. [Google Scholar]
- Zhang, T.N.; Yang, W.; Liu, N. Effect of loop structure of bovine lactoferricin on apoptosis in Jurkat cells. Biometals 2010, 23, 555–561. [Google Scholar]
- Furlong, S.J.; Mader, J.S.; Hoskin, D.W. Lactoferricin-induced apoptosis in estrogen-nonresponsive MDA-MB-435 breast cancer cells is enhanced by C6 ceramide or tamoxifen. Oncol. Rep 2006, 15, 1385–1390. [Google Scholar]
- Yoo, Y.C.; Watanabe, R.; Koike, Y.; Mitobe, M.; Shimazaki, K.; Watanabe, S.; Azuma, I. Apoptosis in human leukemic cells induced by lactoferricin, a bovine milk protein-derived peptide: Involvement of reactive oxygen species. Biochem. Biophys. Res. Commun 1997, 237, 624–628. [Google Scholar]
- Fadnes, B.; Rekdal, O.; Uhlin-Hansen, L. The anticancer activity of lytic peptides is inhibited by heparan sulfate on the surface of the tumor cells. BMC Cancer 2009, 9, 183:1–183:13. [Google Scholar]
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J. Biol. Chem 1988, 263, 16709–16713. [Google Scholar]
- Chen, J.; Xu, X.M.; Underhill, C.B.; Yang, S.; Wang, L.; Chen, Y.; Hong, S.; Creswell, K.; Zhang, L. Tachyplesin activates the classic complement pathway to kill tumor cells. Cancer Res 2005, 65, 4614–4622. [Google Scholar]
- Bullard, K.M.; Kim, H.R.; Wheeler, M.A.; Wilson, C.M.; Neudauer, C.L.; Simpson, M.A.; McCarthy, J.B. Hyaluronan synthase-3 is upregulated in metastatic colon carcinoma cells and manipulation of expression alters matrix retention and cellular growth. Int. J. Cancer 2003, 107, 739–746. [Google Scholar]
- Hautmann, S.H.; Lokeshwar, V.B.; Schroeder, G.L.; Civantos, F.; Duncan, R.C.; Gnann, R.; Friedrich, M.G.; Soloway, M.S. Elevated tissue expression of hyaluronic acid and hyaluronidase validates the HA-HAase urine test for bladder cancer. J. Urol 2001, 165, 2068–2074. [Google Scholar]
- Posey, J.T.; Soloway, M.S.; Ekici, S.; Sofer, M.; Civantos, F.; Duncan, R.C.; Lokeshwar, V.B. Evaluation of the prognostic potential of hyaluronic acid and hyaluronidase (HYAL1) for prostate cancer. Cancer. Res 2003, 63, 2638–2644. [Google Scholar]
- Rooney, P.; Kumar, S.; Ponting, J.; Wang, M. The role of hyaluronan in tumour neovascularization (review). Int. J. Cancer 1995, 60, 632–636. [Google Scholar]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun 1998, 244, 253–257. [Google Scholar]
- Lee, H.S.; Park, C.B.; Kim, J.M.; Jang, S.A.; Park, I.Y.; Kim, M.S.; Cho, J.H.; Kim, S.C. Mechanism of anticancer activity of buforin IIb, a histone H2A-derived peptide. Cancer Lett 2008, 271, 47–55. [Google Scholar]
- Ellerby, H.M.; Bredesen, D.E.; Fujimura, S.; John, V. Hunter-killer peptide (HKP) for targeted therapy. J. Med. Chem 2008, 51, 5887–5892. [Google Scholar]
- Arap, W.; Pasqualini, R. The human vascular mapping project. Selection and utilization of molecules for tumor endothelial targeting. Haemostasis 2001, 31, 30–31. [Google Scholar]
- Souza, B.M.; Mendes, M.A.; Santos, L.D.; Marques, M.R.; Cesar, L.M.; Almeida, R.N.; Pagnocca, F.C.; Konno, K.; Palma, M.S. Structural and functional characterization of two novel peptide toxins isolated from the venom of the social wasp Polybia paulista. Peptides 2005, 26, 2157–2164. [Google Scholar]
- Op den Kamp, J.A. Lipid asymmetry in membranes. Annu. Rev. Biochem 1979, 48, 47–71. [Google Scholar]
- Wang, K.R.; Yan, J.X.; Zhang, B.Z.; Song, J.J.; Jia, P.F.; Wang, R. Novel mode of action of polybia-MPI, a novel antimicrobial peptide, in multi-drug resistant leukemic cells. Cancer Lett 2009, 278, 65–72. [Google Scholar]
- Zhang, W.; Li, J.; Liu, L.W.; Wang, K.R.; Song, J.J.; Yan, J.X.; Li, Z.Y.; Zhang, B.Z.; Wang, R. A novel analog of antimicrobial peptide Polybia-MPI, with thioamide bond substitution, exhibits increased therapeutic efficacy against cancer and diminished toxicity in mice. Peptides 2010, 31, 1832–1838. [Google Scholar]
- Pan, C.Y.; Chen, J.Y.; Cheng, Y.S.; Chen, C.Y.; Ni, I.H.; Sheen, J.F.; Pan, Y.L.; Kuo, C.M. Gene expression and localization of the epinecidin-1 antimicrobial peptide in the grouper (Epinephelus coioides), and its role in protecting fish against pathogenic infection. DNA Cell Biol 2007, 26, 403–413. [Google Scholar]
- Pan, C.Y.; Chen, J.Y.; Ni, I.H.; Wu, J.L.; Kuo, C.M. Organization and promoter analysis of the grouper (Epinephelus coioides) epinecidin-1 gene. Comp. Biochem. Physiol. B Biochem. Mol. Biol 2008, 150, 358–367. [Google Scholar]
- Chen, J.Y.; Lin, W.J.; Wu, J.L.; Her, G.M.; Hui, C.F. Epinecidin-1 peptide induces apoptosis which enhances antitumor effects in human leukemia U937 cells. Peptides 2009, 30, 2365–2373. [Google Scholar]
- Lin, W.J.; Chien, Y.L.; Pan, C.Y.; Lin, T.L.; Chen, J.Y.; Chiu, S.J.; Hui, C.F. Epinecidin-1, an antimicrobial peptide from fish (Epinephelus coioides) which has an antitumor effect like lytic peptides in human fibrosarcoma cells. Peptides 2009, 30, 283–290. [Google Scholar]
- Oren, Z.; Hong, J.; Shai, Y. A repertoire of novel antibacterial diastereomeric peptides with selective cytolytic activity. J. Biol. Chem 1997, 272, 14643–14649. [Google Scholar]
- Makovitzki, A.; Fink, A.; Shai, Y. Suppression of human solid tumor growth in mice by intratumor and systemic inoculation of histidine-rich and pH-dependent host defense-like lytic peptides. Cancer Res 2009, 69, 3458–3463. [Google Scholar]
- Steinstraesser, L.; Hauk, J.; Schubert, C.; Al-Benna, S.; Stricker, I.; Hatt, H.; Shai, Y.; Steinau, H.U.; Jacobsen, F. Suppression of soft tissue sarcoma growth by a host defense-like lytic peptide. PLoS One 2011, 6, e18321:1–e18321:11. [Google Scholar]
- Steinstraesser, L.; Schubert, C.; Hauk, J.; Becerikli, M.; Stricker, I.; Koeller, M.; Hatt, H.; von Duering, M.; Shai, Y.; Steinau, H.U.; et al. Oncolytic designer host defense peptide suppresses growth of human liposarcoma. Int. J. Cancer 2010, 128, 2994–3004. [Google Scholar]
- Saido-Sakanaka, H.; Ishibashi, J.; Momotani, E.; Amano, F.; Yamakawa, M. In vitro and in vivo activity of antimicrobial peptides synthesized based on the insect defensin. Peptides 2004, 25, 19–27. [Google Scholar]
- Saido-Sakanaka, H.; Ishibashi, J.; Momotani, E.; Yamakawa, M. Protective effects of synthetic antibacterial oligopeptides based on the insect defensins on Methicillin-resistant Staphylococcus aureus in mice. Dev. Comp. Immunol 2005, 29, 469–477. [Google Scholar]
- Yamada, M.; Nakamura, K.; Saido-Sakanaka, H.; Asaoka, A.; Yamakawa, M.; Sameshima, T.; Motobu, M.; Hirota, Y. Effect of modified oligopeptides from the beetle Allomyrina dichotoma on Escherichia coli infection in mice. J. Vet. Med. Sci 2004, 66, 137–142. [Google Scholar]
- Yamada, M.; Nakamura, K.; Saido-Sakanaka, H.; Asaoka, A.; Yamakawa, M.; Yamamoto, Y.; Koyama, Y.; Hikosaka, K.; Shimizu, A.; Hirota, Y. Therapeutic effect of modified oligopeptides from the beetle Allomyrina dichotoma on methicillin-resistant Staphylococcus aureus (MRSA) infection in mice. J. Vet. Med. Sci 2005, 67, 1005–1011. [Google Scholar]
- Beschiaschvili, G.; Seelig, J. Melittin binding to mixed phosphatidylglycerol/phosphatidylcholine membranes. Biochemistry 1990, 29, 52–58. [Google Scholar]
- Marsh, D. Peptide models for membrane channels. Biochem. J 1996, 315, 345–361. [Google Scholar]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 1995, 34, 6521–6526. [Google Scholar]
- Pawlak, M.; Kuhn, A.; Vogel, H. Pf3 coat protein forms voltage-gated ion channels in planar lipid bilayers. Biochemistry 1994, 33, 283–290. [Google Scholar]
- Schwarz, G.; Gerke, H.; Rizzo, V.; Stankowski, S. Incorporation kinetics in a membrane, studied with the pore-forming peptide alamethicin. Biophys. J 1987, 52, 685–692. [Google Scholar]
- Vogel, H.; Jahnig, F. The structure of melittin in membranes. Biophys. J 1986, 50, 573–582. [Google Scholar]
- Ladokhin, A.S.; White, S.H. “Detergent-like” permeabilization of anionic lipid vesicles by melittin. Biochim. Biophys. Acta 2001, 1514, 253–260. [Google Scholar]
- Oren, Z.; Shai, Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers 1998, 47, 451–463. [Google Scholar]
- Doyle, J.; Brinkworth, C.S.; Wegener, K.L.; Carver, J.A.; Llewellyn, L.E.; Olver, I.N.; Bowie, J.H.; Wabnitz, P.A.; Tyler, M.J. nNOS inhibition, antimicrobial and anticancer activity of the amphibian skin peptide, citropin 1.1 and synthetic modifications. The solution structure of a modified citropin 1.1. Eur. J. Biochem 2003, 270, 1141–1153. [Google Scholar]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar]


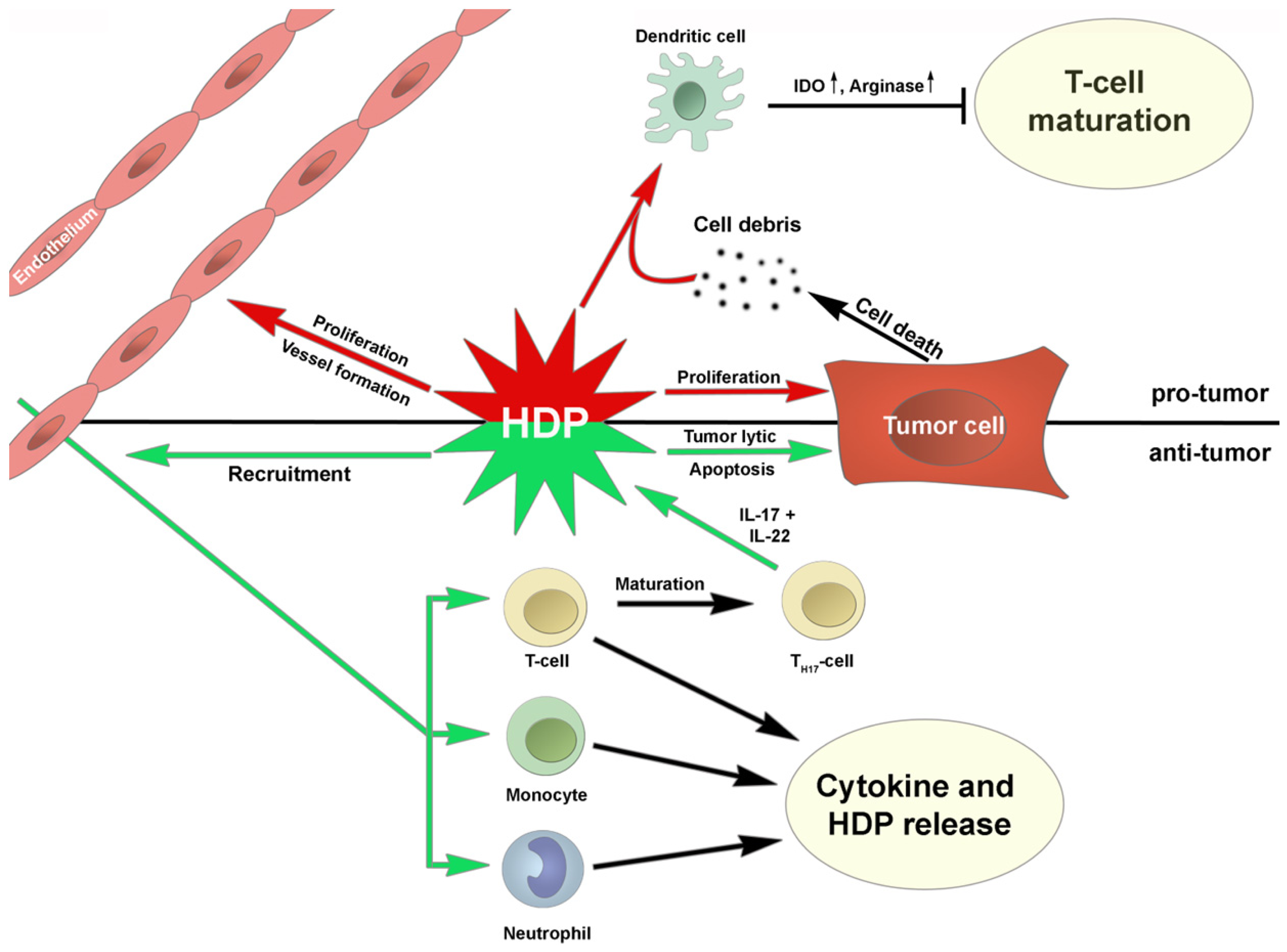{kind=link}
| Peptide | Cancer cell | Study | Reference |
|---|---|---|---|
| Polybia-MPI | Human bladder and prostate | In vitro, compared against murine fibroblasts | Wang et al. 2008 |
| Tachyplesin | Prostate cancer cell line | In vitro and syngenic mouse model | Chen et al. 2001 |
| Buforin IIb | 62 different cancer cell lines | In vivo mouse xenograft model | |
| Human breast cancer cell line | In vitro | Furlong et al. 2006 | |
| Human neuroblastoma cell lines | In vitro and neuroblastoma mouse xenograft model | Eliassen et al. 2006 | |
| Bovine lactoferricin | Jurkat T-leukemia cells | In vitro | Mader et al. 2007 |
| adenocarcinoma | In vivo tumor induced F344 rats | Tsuda et al. 1998 | |
| Human monocytic leukemic cells | In vitro | Yoo et al. 1997 | |
| Cecropin A and B | Bladder cancer cells | In vitro | Suttmann et al. 2008 |
| Human alpha-defensin-1 | Human lung adenocarcinoma | Mouse xenograft model | Xu et al. 2008 |
| Human alpha-defensin-1 and lactoferrin | oral squamous cell carcinoma | In vitro | McKeown et al. 2006 |
| Human beta-defensin-1 | Prostate cancer cell lines | In vitro | Bullard et al. 2008 |
| hCAP-18(109–135) | oral squamous cell carcinoma | In vitro | Okumura et al. 2004 |
| Magainin II | Bladder cancer cell line hematopoietic tumor and solid tumor target cells | In vitro | Lehmann et al. 2006 |
| In vitro | Cruciani et al. 1991 | ||
| Magainin II and derivatives | Several cell lines | In vivo ovarian cancer model | Baker et al. 1993 |
| MSI-511 (Magainin derivative) | Several human melanoma cell lines | In vitro | Soballe et al. 1995 |
| Magainin A and G | Small cell lung cancer cell lines | In vitro | Ohsaki et al. 1992 |
| Sythetic 9-mer d-peptides | Mouse myeloma cell line | In vitro | Iwasaki et al. 2009 |
| CA-ME (hybrid peptide) | Small cell lung cancer cell lines | In vitro | Shin et al. 1997 |
| Prostate carcinoma cell lines | Mouse xenograft model | Papo et al. 2004 | |
| Synthetic l/d-peptides | Primary human prostate and breast cancer cells | Prevents metastases in mouse xenograft model | Papo et al. 2006 |
| Mouse melanoma and lung cancer cell lines | Mouse xenograft model | Papo et al. 2003 | |
| Human liposarcoma and synovial sarcoma cells | Mouse xenograft model | Steinstraeser et al. 2011 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Benna, S.; Shai, Y.; Jacobsen, F.; Steinstraesser, L. Oncolytic Activities of Host Defense Peptides. Int. J. Mol. Sci. 2011, 12, 8027-8051. https://doi.org/10.3390/ijms12118027
Al-Benna S, Shai Y, Jacobsen F, Steinstraesser L. Oncolytic Activities of Host Defense Peptides. International Journal of Molecular Sciences. 2011; 12(11):8027-8051. https://doi.org/10.3390/ijms12118027
Chicago/Turabian StyleAl-Benna, Sammy, Yechiel Shai, Frank Jacobsen, and Lars Steinstraesser. 2011. "Oncolytic Activities of Host Defense Peptides" International Journal of Molecular Sciences 12, no. 11: 8027-8051. https://doi.org/10.3390/ijms12118027