Photodynamic Treatment Induces an Apoptotic Pathway Involving Calcium, Nitric Oxide, p53, p21-Activated Kinase 2, and c-Jun N-Terminal Kinase and Inactivates Survival Signal in Human Umbilical Vein Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
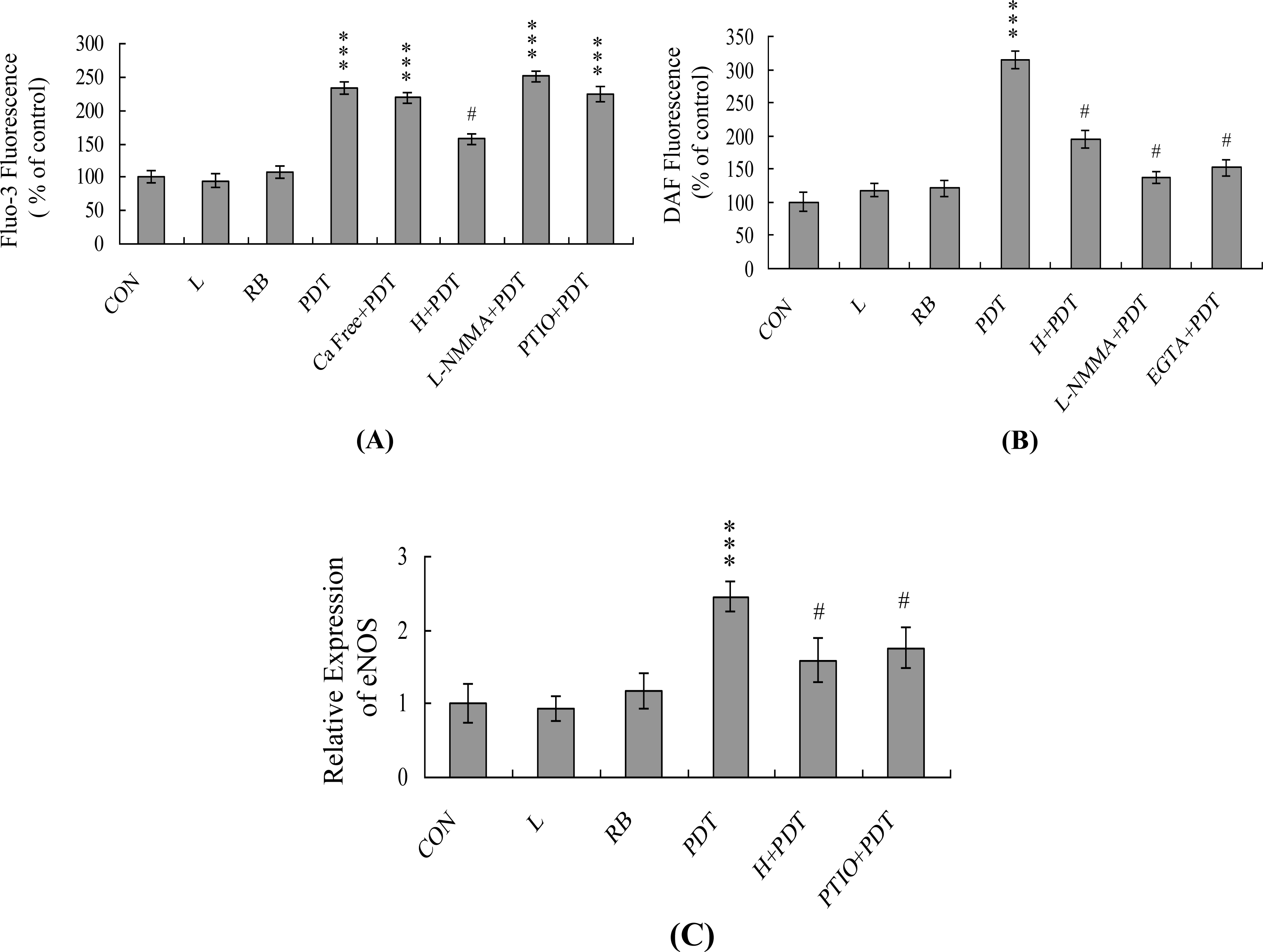
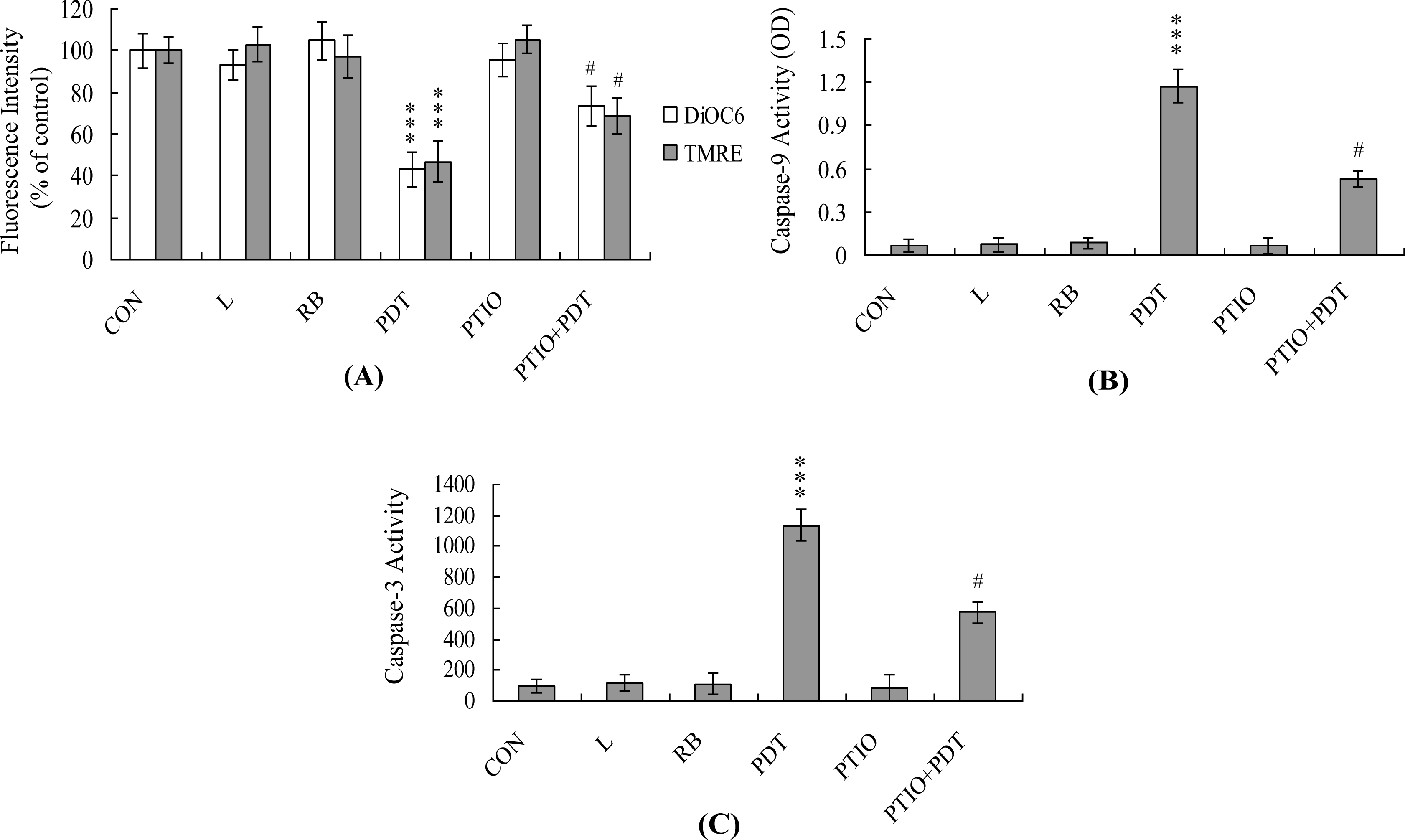
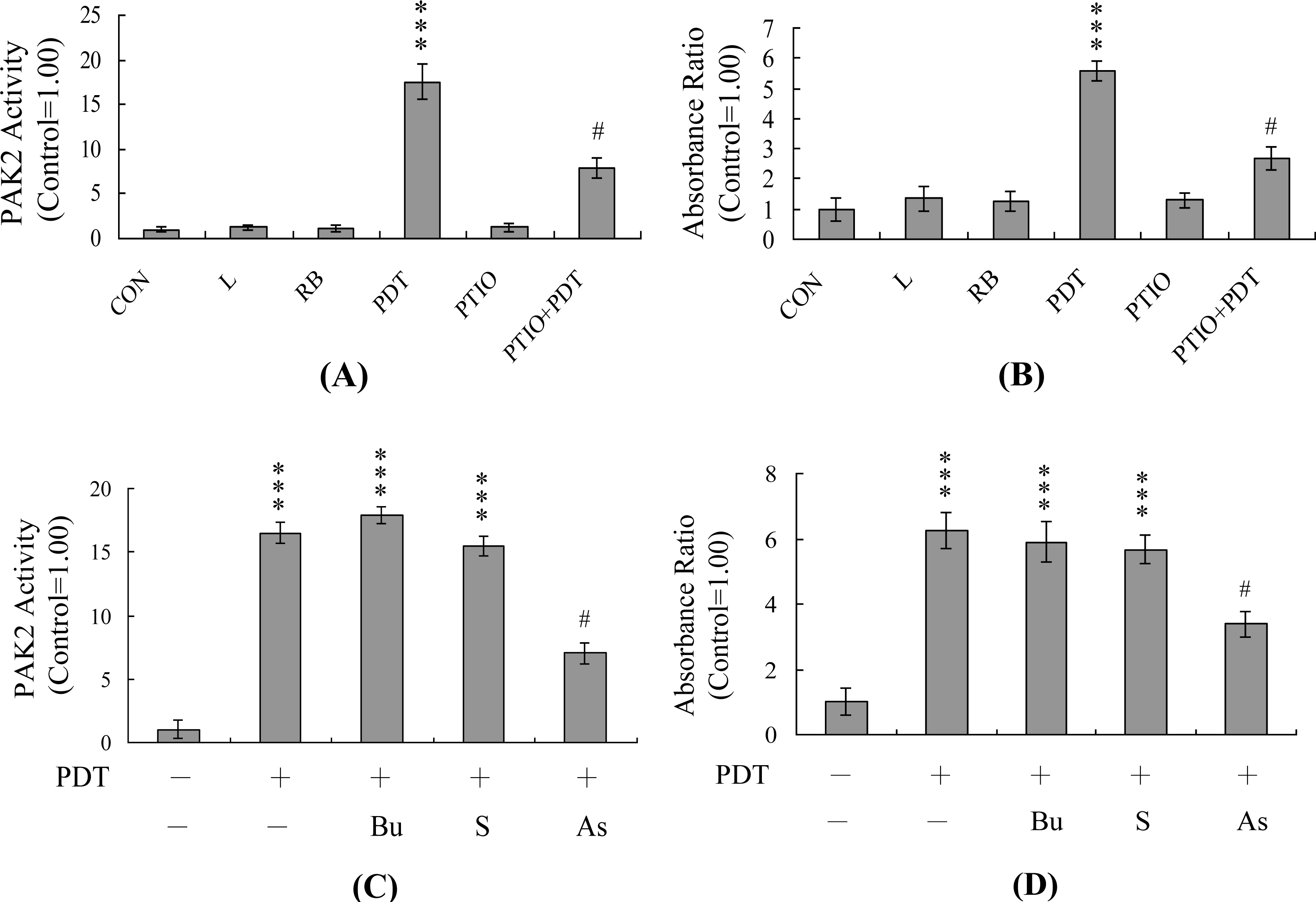
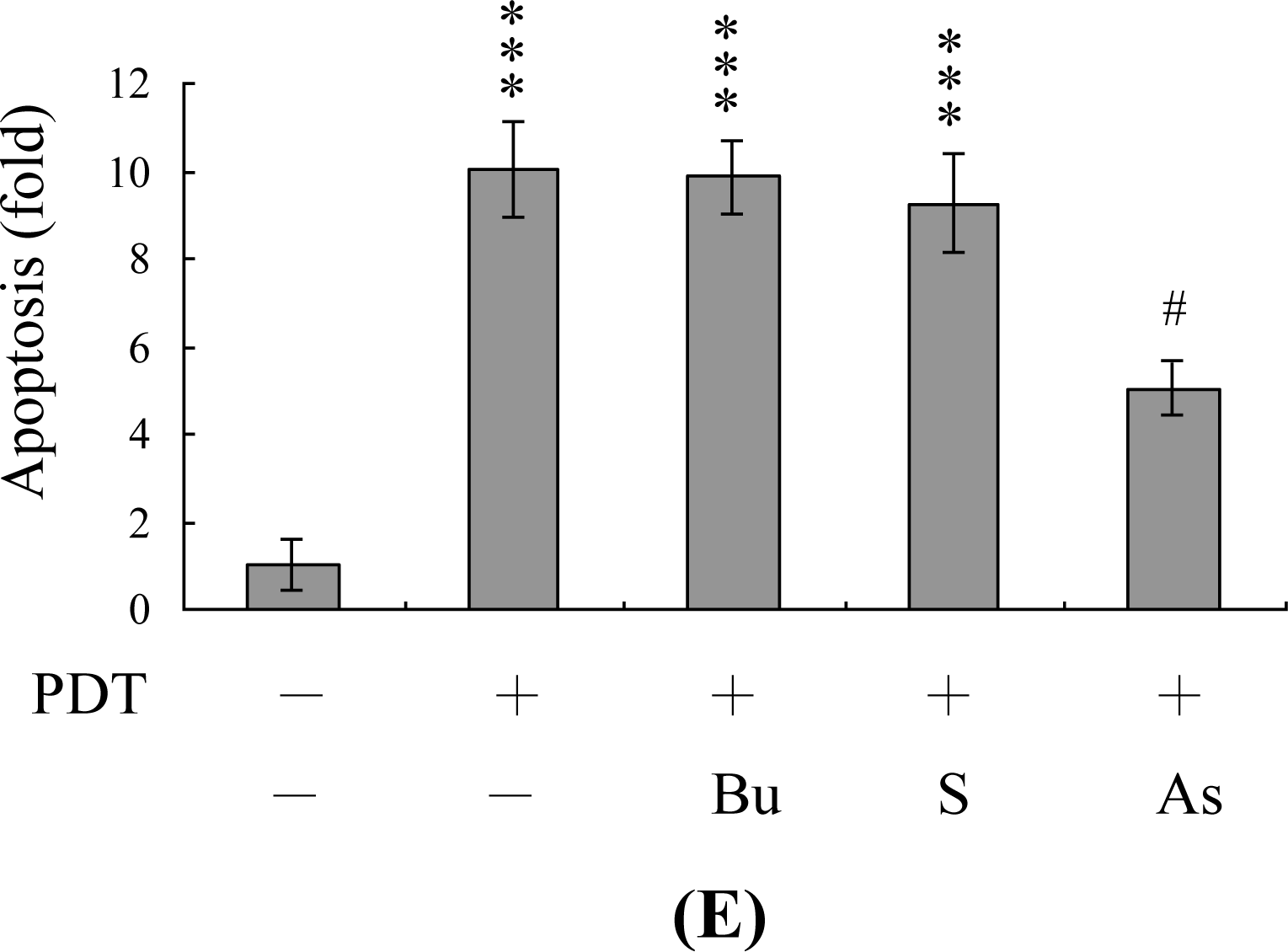
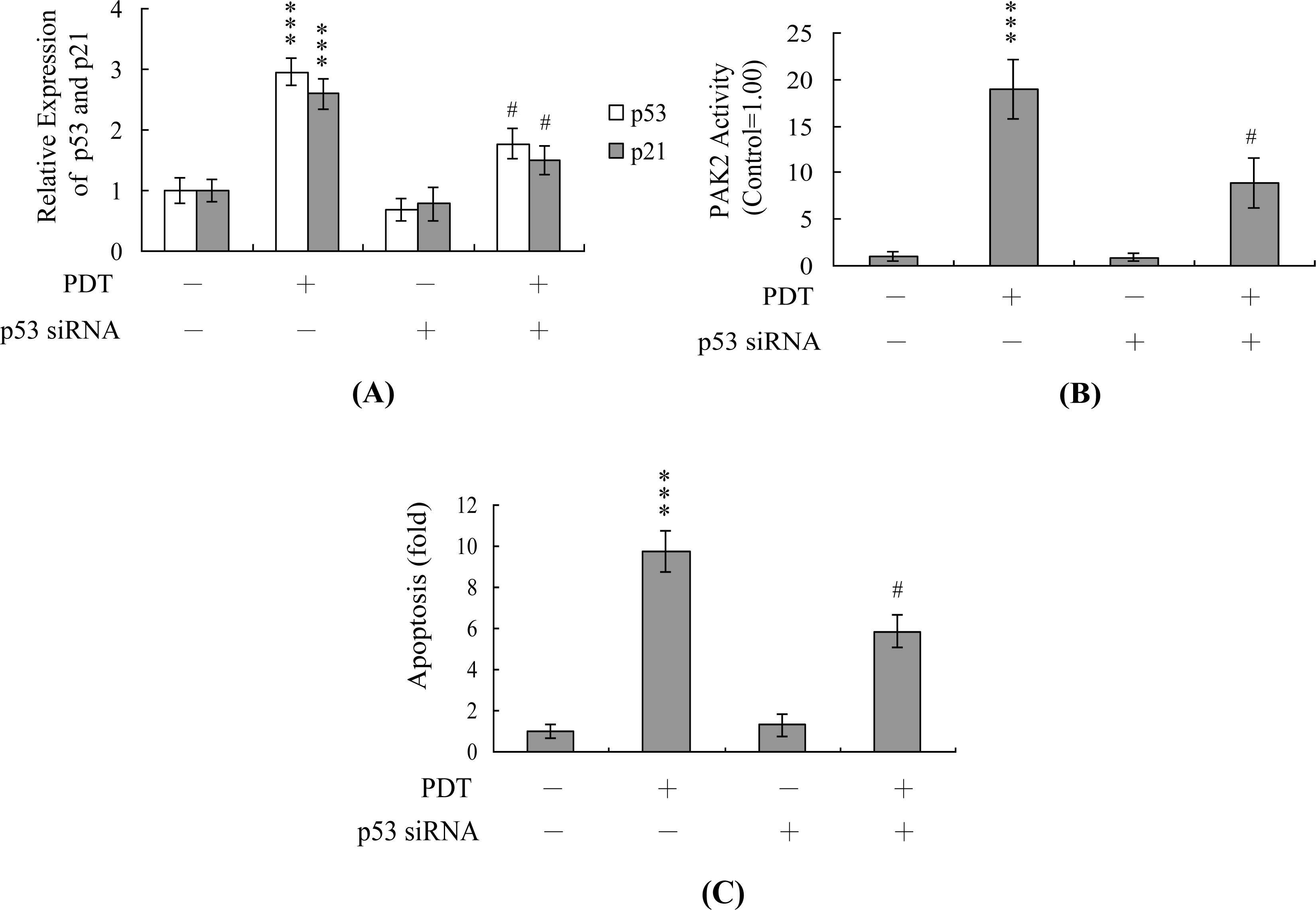
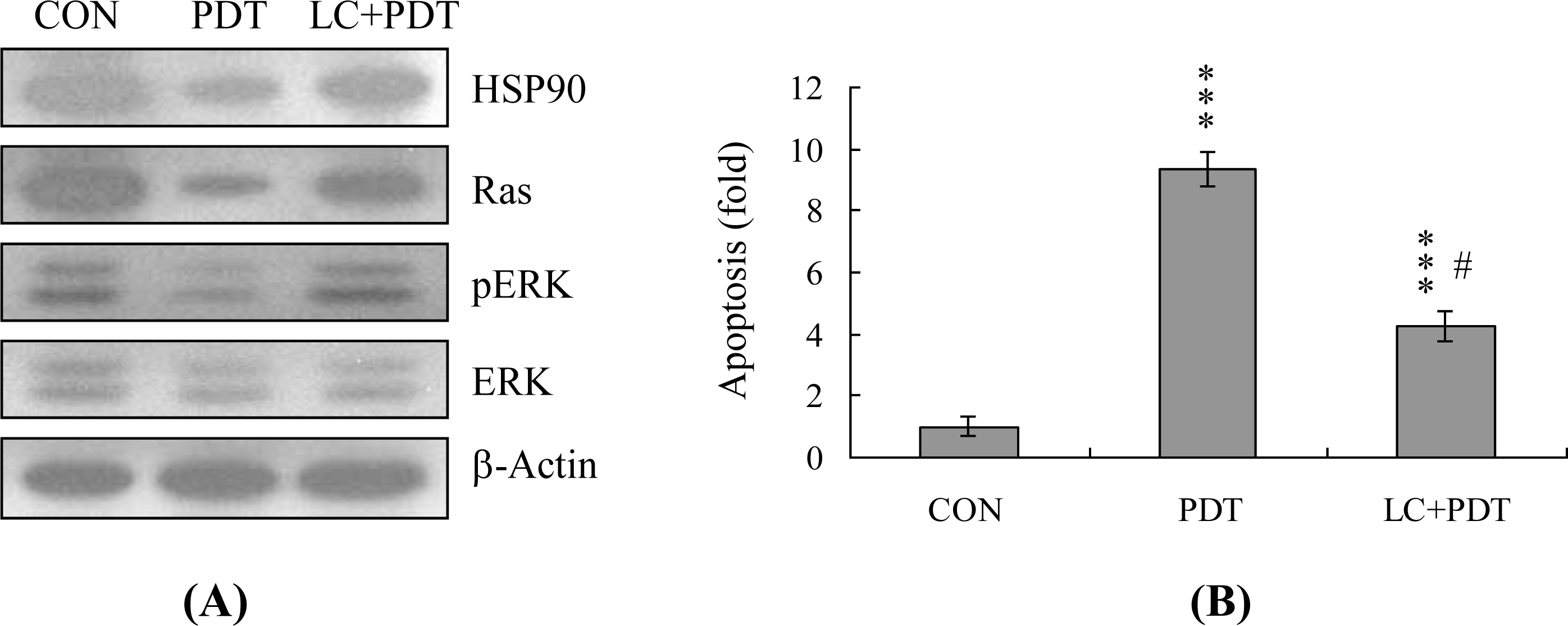
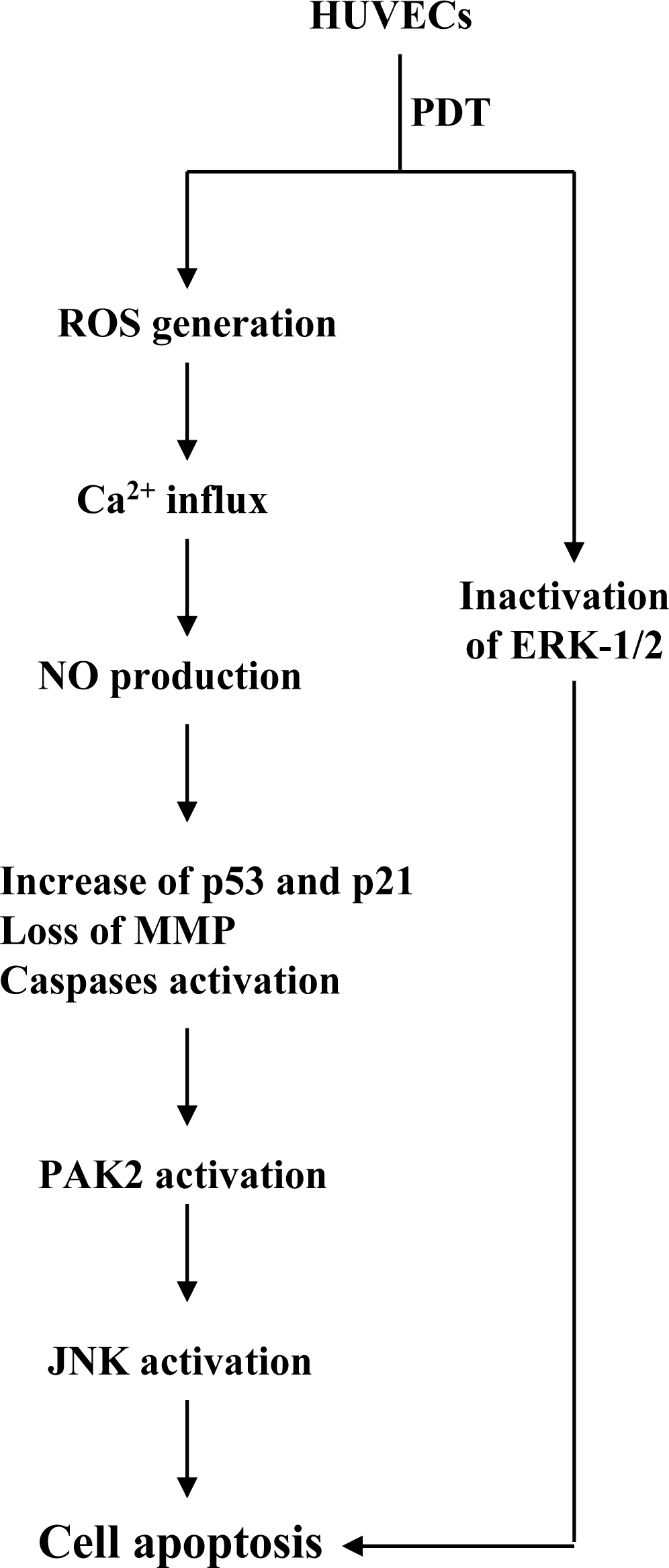
2. Results and Discussion
3. Experimental Section
3.1. Chemicals
3.2. Cell Culture and PDT
3.3. Apoptosis Assay
3.4. ROS Assay
3.5. Detection of Intracellular Calcium Concentration ([Ca2+]i)
3.6. Detection of Intracellular NO Content
3.7. Detection of Mitochondrial Membrane Potential (MMP)
3.8. Caspase Activity Assays
3.9. Immunoprecipitation and PAK2 Activity Assay
3.10. JNK Assays
3.11. Inhibition of PAK2 by Anti-sense Oligonucleotides
3.12. Real-Time RT-PCR Assay
3.13. siRNA Knockdown
3.14. Immunoblots
3.15. Statistics
4. Conclusions
Acknowledgments
References
- Girotti, A. Photodynamic Therapy of Neoplastic Disease; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Henderson, BW; Dougherty, TJ. How does photodynamic therapy work? Photochem. Photobiol 1992, 55, 145–157. [Google Scholar]
- Halliwell, B; Gutteridge, JMC. Free Radicals in Biology and Medicine; Oxford University: New York, NY, USA, 1989. [Google Scholar]
- Johnson, BE; Ferguson, J. Drug and chemical photosensitivity. Semin. Dermatol 1990, 9, 39–46. [Google Scholar]
- Pass, HI. Photodynamic therapy in oncology: mechanisms and clinical use. J. Natl. Cancer Inst 1993, 85, 443–456. [Google Scholar]
- Dougherty, TJ; Gomer, CJ; Henderson, BW; Jori, G; Kessel, D; Korbelik, M; Moan, J; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst 1998, 90, 889–905. [Google Scholar]
- Agarwal, ML; Clay, ME; Harvey, EJ; Evans, HH; Antunez, AR; Oleinick, NL. Photodynamic therapy induces rapid cell death by apoptosis in L5178Y mouse lymphoma cells. Cancer Res 1991, 51, 5993–5996. [Google Scholar]
- Thompson, CB. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar]
- Chan, WH; Yu, JS; Yang, SD. Apoptotic signalling cascade in photosensitized human epidermal carcinoma A431 cells: involvement of singlet oxygen, c-Jun N-terminal kinase, caspase-3 and p21-activated kinase 2. Biochem. J 2000, 351, 221–232. [Google Scholar]
- Chan, WH; Wu, HJ. Anti-apoptotic effects of curcumin on photosensitized human epidermal carcinoma A431 cells. J. Cell. Biochem 2004, 92, 200–212. [Google Scholar]
- Ekmekcioglu, S; Tang, CH; Grimm, EA. NO news is not necessarily good news in cancer. Curr. Cancer Drug Targets 2005, 5, 103–115. [Google Scholar]
- Zhou, J; Brune, B. NO and transcriptional regulation: from signaling to death. Toxicology 2005, 208, 223–233. [Google Scholar]
- Rao, CV. Nitric oxide signaling in colon cancer chemoprevention. Mutat. Res 2004, 555, 107–119. [Google Scholar]
- Lu, Z; Tao, Y; Zhou, Z; Zhang, J; Li, C; Ou, L; Zhao, B. Mitochondrial reactive oxygen species and nitric oxide-mediated cancer cell apoptosis in 2-butylamino-2-demethoxyhypocrellin B photodynamic treatment. Free Radic. Biol. Med 2006, 41, 1590–1605. [Google Scholar]
- Nazarewicz, RR; Zenebe, WJ; Parihar, A; Larson, SK; Alidema, E; Choi, J; Ghafourifar, P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res 2007, 67, 1282–1290. [Google Scholar]
- Ghafourifar, P; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci 2005, 26, 190–195. [Google Scholar]
- Brookes, PS. Mitochondrial nitric oxide synthase. Mitochondrion 2004, 3, 187–204. [Google Scholar]
- Dennis, J; Bennett, JP, Jr. Interactions among nitric oxide and Bcl-family proteins after MPP+ exposure of SH-SY5Y neural cells I: MPP+ increases mitochondrial NO and Bax protein. J. Neurosci. Res 2003, 72, 76–88. [Google Scholar]
- Elfering, SL; Sarkela, TM; Giulivi, C. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem 2002, 277, 38079–38086. [Google Scholar]
- Dedkova, EN; Ji, X; Lipsius, SL; Blatter, LA. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol 2004, 286, C406–C415. [Google Scholar]
- Anderson, P. Kinase cascades regulating entry into apoptosis. Microbiol. Mol. Biol. Rev 1997, 61, 33–46. [Google Scholar]
- Xia, Z; Dickens, M; Raingeaud, J; Davis, RJ; Greenberg, ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar]
- Verheij, M; Bose, R; Lin, XH; Yao, B; Jarvis, WD; Grant, S; Birrer, MJ; Szabo, E; Zon, LI; Kyriakis, JM; Haimovitz-Friedman, A; Fuks, Z; Kolesnick, RN. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 1996, 380, 75–79. [Google Scholar]
- Seimiya, H; Mashima, T; Toho, M; Tsuruo, T. c-Jun NH2-terminal kinase-mediated activation of interleukin-1beta converting enzyme/CED-3-like protease during anticancer drug-induced apoptosis. J. Biol. Chem 1997, 272, 4631–4636. [Google Scholar]
- Rudel, T; Bokoch, GM. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science 1997, 276, 1571–1574. [Google Scholar]
- Lee, N; MacDonald, H; Reinhard, C; Halenbeck, R; Roulston, A; Shi, T; Williams, LT. Activation of hPAK65 by caspase cleavage induces some of the morphological and biochemical changes of apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 13642–13647. [Google Scholar]
- Tang, TK; Chang, WC; Chan, WH; Yang, SD; Ni, MH; Yu, JS. Proteolytic cleavage and activation of PAK2 during UV irradiation-induced apoptosis in A431 cells. J. Cell. Biochem 1998, 70, 442–454. [Google Scholar]
- Chan, WH; Yu, JS; Yang, SD. PAK2 is cleaved and activated during hyperosmotic shock-induced apoptosis via a caspase-dependent mechanism: evidence for the involvement of oxidative stress. J. Cell. Physiol 1999, 178, 397–408. [Google Scholar]
- Zhang, S; Han, J; Sells, MA; Chernoff, J; Knaus, UG; Ulevitch, RJ; Bokoch, GM. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J. Biol. Chem 1995, 270, 23934–23936. [Google Scholar]
- Frost, JA; Xu, S; Hutchison, MR; Marcus, S; Cobb, MH. Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol. Cell. Biol 1996, 16, 3707–3713. [Google Scholar]
- Blagosklonny, MV. Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia 2002, 16, 455–462. [Google Scholar]
- Caraglia, M; Abbruzzese, A; Leardi, A; Pepe, S; Budillon, A; Baldassare, G; Selleri, C; Lorenzo, SD; Fabbrocini, A; Giuberti, G; Vitale, G; Lupoli, G; Bianco, AR; Tagliaferri, P. Interferon-alpha induces apoptosis in human KB cells through a stress-dependent mitogen activated protein kinase pathway that is antagonized by epidermal growth factor. Cell Death Differ 1999, 6, 773–780. [Google Scholar]
- Tao, J; Sanghera, JS; Pelech, SL; Wong, G; Levy, JG. Stimulation of stress-activated protein kinase and p38 HOG1 kinase in murine keratinocytes following photodynamic therapy with benzoporphyrin derivative. J. Biol. Chem 1996, 271, 27107–27115. [Google Scholar]
- Klotz, LO; Briviba, K; Sies, H. Singlet oxygen mediates the activation of JNK by UVA radiation in human skin fibroblasts. FEBS Lett 1997, 408, 289–291. [Google Scholar]
- Basu-Modak, S; Tyrrell, RM. Singlet oxygen: a primary effector in the ultraviolet A/near-visible light induction of the human heme oxygenase gene. Cancer Res 1993, 53, 4505–4510. [Google Scholar]
- Li, P; Nijhawan, D; Budihardjo, I; Srinivasula, SM; Ahmad, M; Alnemri, ES; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar]
- Zou, H; Henzel, WJ; Liu, X; Lutschg, A; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar]
- Weber, T; Dalen, H; Andera, L; Negre-Salvayre, A; Auge, N; Sticha, M; Lloret, A; Terman, A; Witting, PK; Higuchi, M; Plasilova, M; Zivny, J; Gellert, N; Weber, C; Neuzil, J. Mitochondria play a central role in apoptosis induced by alpha-tocopheryl succinate, an agent with antineoplastic activity: comparison with receptor-mediated pro-apoptotic signaling. Biochemistry 2003, 42, 4277–4291. [Google Scholar]
- Chiu, SM; Oleinick, NL. Dissociation of mitochondrial depolarization from cytochrome c release during apoptosis induced by photodynamic therapy. Br. J. Cancer 2001, 84, 1099–1106. [Google Scholar]
- Caraglia, M; Tagliaferri, P; Marra, M; Giuberti, G; Budillon, A; Gennaro, ED; Pepe, S; Vitale, G; Improta, S; Tassone, P; Venuta, S; Bianco, AR; Abbruzzese, A. EGF activates an inducible survival response via the RAS→Erk-1/2 pathway to counteract interferon-alpha-mediated apoptosis in epidermoid cancer cells. Cell Death Differ 2003, 10, 218–229. [Google Scholar]
- Caraglia, M; Marra, M; Pelaia, G; Maselli, R; Caputi, M; Marsico, SA; Abbruzzese, A. Alpha-interferon and its effects on signal transduction pathways. J. Cell. Physiol 2005, 202, 323–335. [Google Scholar]
- Chan, WH; Shiao, NH; Lu, PZ. CdSe quantum dots induce apoptosis in human neuroblastoma cells via mitochondrial-dependent pathways and inhibition of survival signals. Toxicol. Lett 2006, 167, 191–200. [Google Scholar]
- Chan, WH. Citrinin induces apoptosis via a mitochondria-dependent pathway and inhibition of survival signals in embryonic stem cells, and causes developmental injury in blastocysts. Biochem. J 2007, 404, 317–326. [Google Scholar]
- Almeida, RD; Manadas, BJ; Carvalho, AP; Duarte, CB. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 2004, 1704, 59–86. [Google Scholar]
- Inanami, O; Yoshito, A; Takahashi, K; Hiraoka, W; Kuwabara, M. Effects of BAPTA-AM and forskolin on apoptosis and cytochrome c release in photosensitized Chinese hamster V79 cells. Photochem. Photobiol 1999, 70, 650–655. [Google Scholar]
- Monteiro, HP; Silva, EF; Stern, A. Nitric oxide: a potential inducer of adhesion-related apoptosis—anoikis. Nitric Oxide 2004, 10, 1–10. [Google Scholar]
- Li, CQ; Wogan, GN. Nitric oxide as a modulator of apoptosis. Cancer Lett 2005, 226, 1–15. [Google Scholar]
- Gomes, ER; Almeida, RD; Carvalho, AP; Duarte, CB. Nitric oxide modulates tumor cell death induced by photodynamic therapy through a cGMP-dependent mechanism. Photochem. Photobiol 2002, 76, 423–430. [Google Scholar]
- Chan, WH; Yu, JS; Yang, SD. Heat shock stress induces cleavage and activation of PAK2 in apoptotic cells. J. Protein Chem 1998, 17, 485–494. [Google Scholar]
- Rooney, RD; Tuazon, PT; Meek, WE; Carroll, EJ; Hagen, JJ; Gump, EL; Monnig, CA; Lugo, T; Traugh, JA. Cleavage arrest of early frog embryos by the G protein-activated protein kinase PAK I. J. Biol. Chem 1996, 271, 21498–21504. [Google Scholar]
- Faure, S; Vigneron, S; Doree, M; Morin, N. A member of the Ste20/PAK family of protein kinases is involved in both arrest of Xenopus oocytes at G2/prophase of the first meiotic cell cycle and in prevention of apoptosis. EMBO J 1997, 16, 5550–5561. [Google Scholar]
- Tsai, IC; Hsieh, YJ; Lyu, PC; Yu, JS. Anti-phosphopeptide antibody, P-STM as a novel tool for detecting mitotic phosphoproteins: identification of lamins A and C as two major targets. J. Cell. Biochem 2005, 94, 967–981. [Google Scholar]
- Chan, WH; Wu, HJ; Shiao, NH. Apoptotic signaling in methylglyoxal-treated human osteoblasts involves oxidative stress, c-Jun N-terminal kinase, caspase-3, and p21-activated kinase 2. J. Cell. Biochem 2007, 100, 1056–1069. [Google Scholar]
- Li, CQ; Robles, AI; Hanigan, CL; Hofseth, LJ; Trudel, LJ; Harris, CC; Wogan, GN. Apoptotic signaling pathways induced by nitric oxide in human lymphoblastoid cells expressing wild-type or mutant p53. Cancer Res 2004, 64, 3022–3029. [Google Scholar]
- Okada, H; Mak, TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar]
- French, PW; Penny, R; Laurence, JA; McKenzie, DR. Mobile phones, heat shock proteins and cancer. Differentiation 2001, 67, 93–97. [Google Scholar]
- Leszczynski, D; Joenvaara, S; Reivinen, J; Kuokka, R. Non-thermal activation of the hsp27/p38MAPK stress pathway by mobile phone radiation in human endothelial cells: molecular mechanism for cancer- and blood-brain barrier-related effects. Differentiation 2002, 70, 120–129. [Google Scholar]
- Mayer, MP; Bukau, B. Molecular chaperones: the busy life of Hsp90. Curr. Biol 1999, 9, R322–R325. [Google Scholar]
- Bellyei, S; Szigeti, A; Boronkai, A; Pozsgai, E; Gomori, E; Melegh, B; Janaky, T; Bognar, Z; Hocsak, E; Sumegi, B; Gallyas, F, Jr. Inhibition of cell death by a novel 16.2 kD heat shock protein predominantly via Hsp90 mediated lipid rafts stabilization and Akt activation pathway. Apoptosis 2007, 12, 97–112. [Google Scholar]
- Aoshima, H; Satoh, T; Sakai, N; Yamada, M; Enokido, Y; Ikeuchi, T; Hatanaka, H. Generation of free radicals during lipid hydroperoxide-triggered apoptosis in PC12h cells. Biochim. Biophys. Acta 1997, 1345, 35–42. [Google Scholar]
- Nakatsubo, N; Kojima, H; Kikuchi, K; Nagoshi, H; Hirata, Y; Maeda, D; Imai, Y; Irimura, T; Nagano, T. Direct evidence of nitric oxide production from bovine aortic endothelial cells using new fluorescence indicators: diaminofluoresceins. FEBS Lett 1998, 427, 263–266. [Google Scholar]
- Chan, WH; Wu, CC; Yu, JS. Curcumin inhibits UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermoid carcinoma A431 cells. J. Cell. Biochem 2003, 90, 327–338. [Google Scholar]
- Hsieh, YJ; Wu, CC; Chang, CJ; Yu, JS. Subcellular localization of Photofrin determines the death phenotype of human epidermoid carcinoma A431 cells triggered by photodynamic therapy: when plasma membranes are the main targets. J. Cell. Physiol 2003, 194, 363–375. [Google Scholar]
- Huang, YT; Lai, CY; Lou, SL; Yeh, JM; Chan, WH. Activation of JNK and PAK2 is essential for citrinin-induced apoptosis in a human osteoblast cell line. Environ. Toxicol 2009, 24, 343–356. [Google Scholar]
- Jakobi, R; Chen, CJ; Tuazon, PT; Traugh, JA. Molecular cloning and sequencing of the cytostatic G protein-activated protein kinase PAK I. J. Biol. Chem 1996, 271, 6206–6211. [Google Scholar]
- Martin, GA; Bollag, G; McCormick, F; Abo, A. A novel serine kinase activated by rac1/CDC42Hs-dependent autophosphorylation is related to PAK65 and STE20. EMBO J 1995, 14, 1970–1978. [Google Scholar]
- Reichlin, M. Use of glutaraldehyde as a coupling agent for proteins and peptides. Methods Enzymol 1980, 70, 159–165. [Google Scholar]
- Reimann, EM; Walsh, DA; Krebs, EG. Purification and properties of rabbit skeletal muscle adenosine 3′,5′-monophosphate-dependent protein kinases. J. Biol. Chem 1971, 246, 1986–1995. [Google Scholar]
- Lu, PZ; Lai, CY; Chan, WH. Caffeine Induces Cell Death via Activation of Apoptotic Signal and Inactivation of Survival Signal in Human Osteoblasts. Int. J. Mol. Sci 2008, 9, 698–718. [Google Scholar]









© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chan, W.-H. Photodynamic Treatment Induces an Apoptotic Pathway Involving Calcium, Nitric Oxide, p53, p21-Activated Kinase 2, and c-Jun N-Terminal Kinase and Inactivates Survival Signal in Human Umbilical Vein Endothelial Cells. Int. J. Mol. Sci. 2011, 12, 1041-1059. https://doi.org/10.3390/ijms12021041
Chan W-H. Photodynamic Treatment Induces an Apoptotic Pathway Involving Calcium, Nitric Oxide, p53, p21-Activated Kinase 2, and c-Jun N-Terminal Kinase and Inactivates Survival Signal in Human Umbilical Vein Endothelial Cells. International Journal of Molecular Sciences. 2011; 12(2):1041-1059. https://doi.org/10.3390/ijms12021041
Chicago/Turabian StyleChan, Wen-Hsiung. 2011. "Photodynamic Treatment Induces an Apoptotic Pathway Involving Calcium, Nitric Oxide, p53, p21-Activated Kinase 2, and c-Jun N-Terminal Kinase and Inactivates Survival Signal in Human Umbilical Vein Endothelial Cells" International Journal of Molecular Sciences 12, no. 2: 1041-1059. https://doi.org/10.3390/ijms12021041