Sulfotyrosine Recognition as Marker for Druggable Sites in the Extracellular Space

Abstract
:1. Introduction
2. Methods
2.1. Chemokine Multiple-Sequence Alignment
2.2. Chemokine Structures and Models
2.3. Structural Homology Modeling
2.4. Protein Expression and Purification
2.5. NMR Analysis
3. Results and Discussion
3.1. Chemokine Primary Sequence Alignment
3.2. Homology Modeling
3.3. Structural Alignment
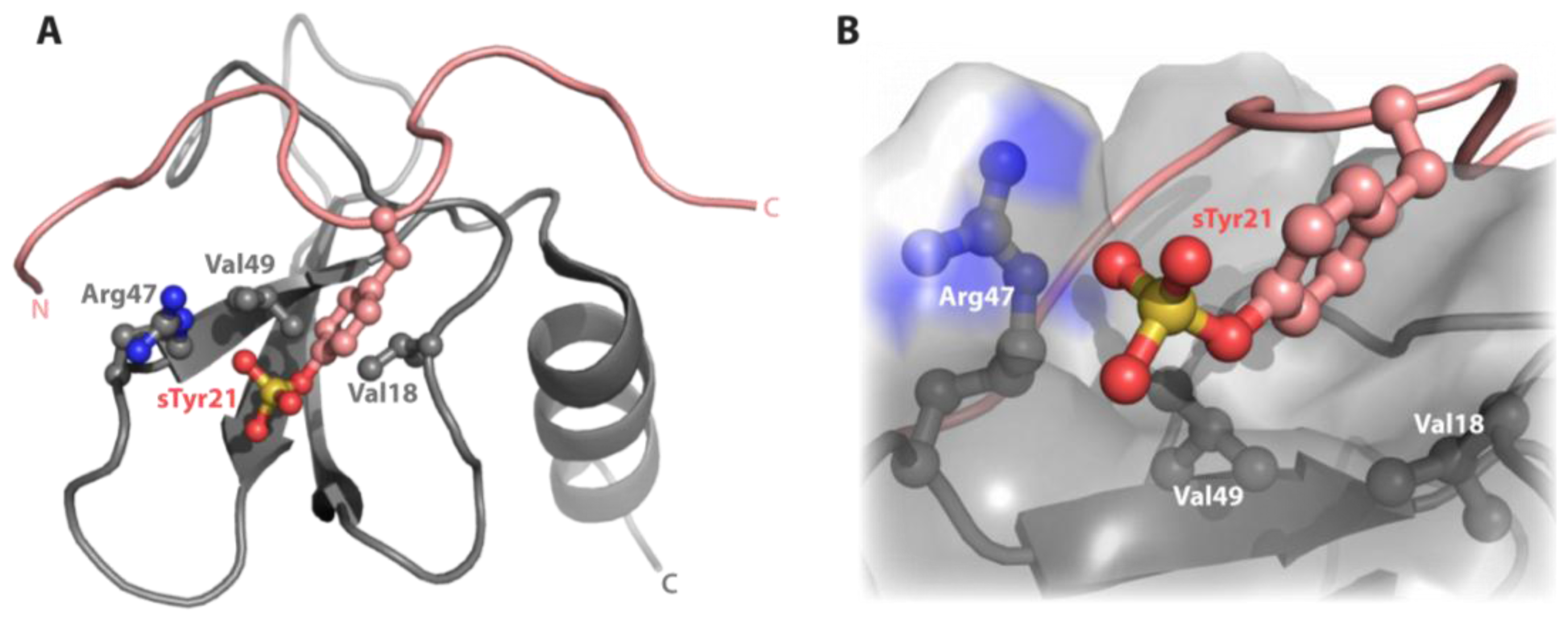
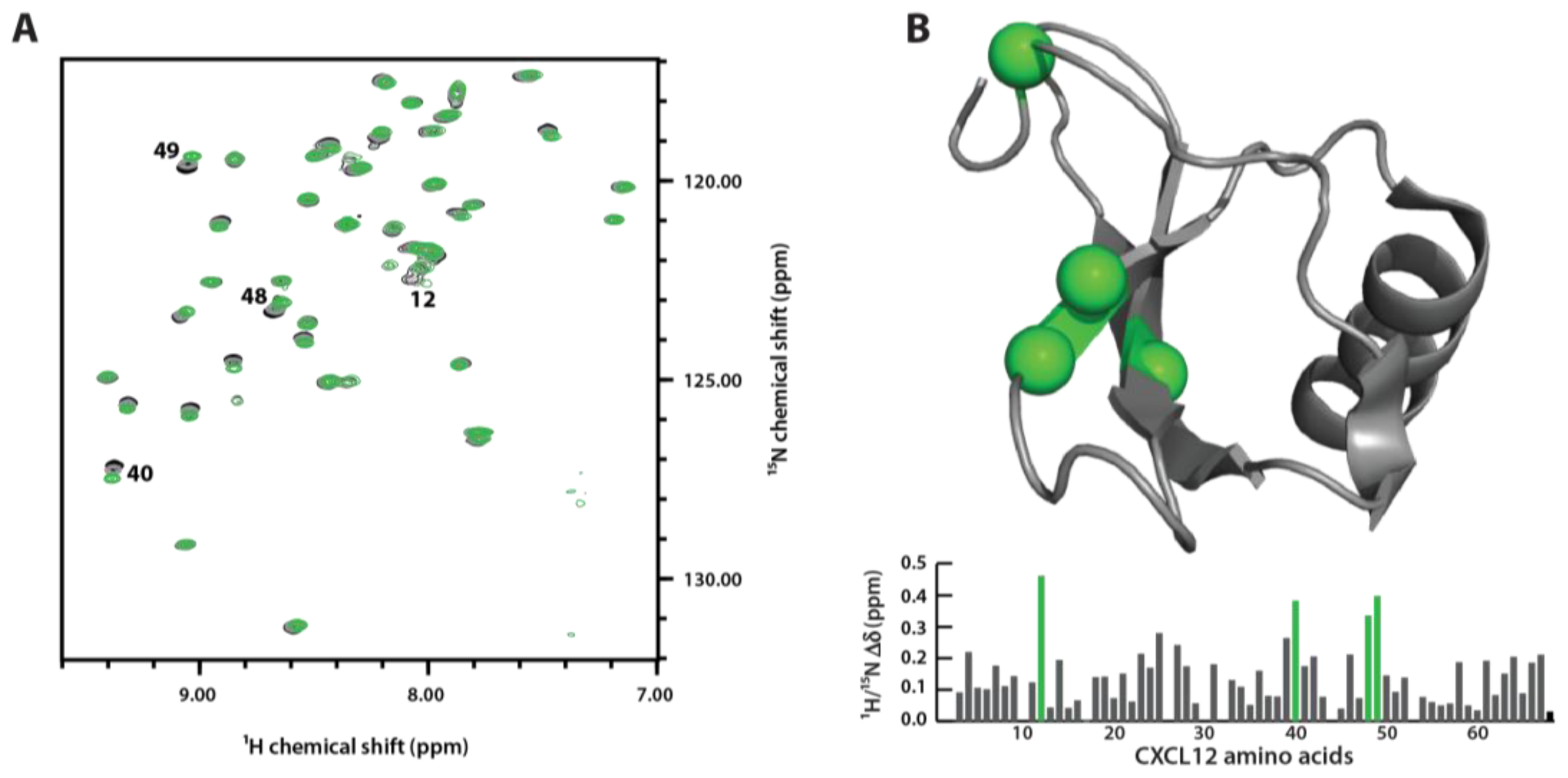
3.4. Sulfotyrosine Titration Identifies CXCL12 sTyr21 Binding Pocket
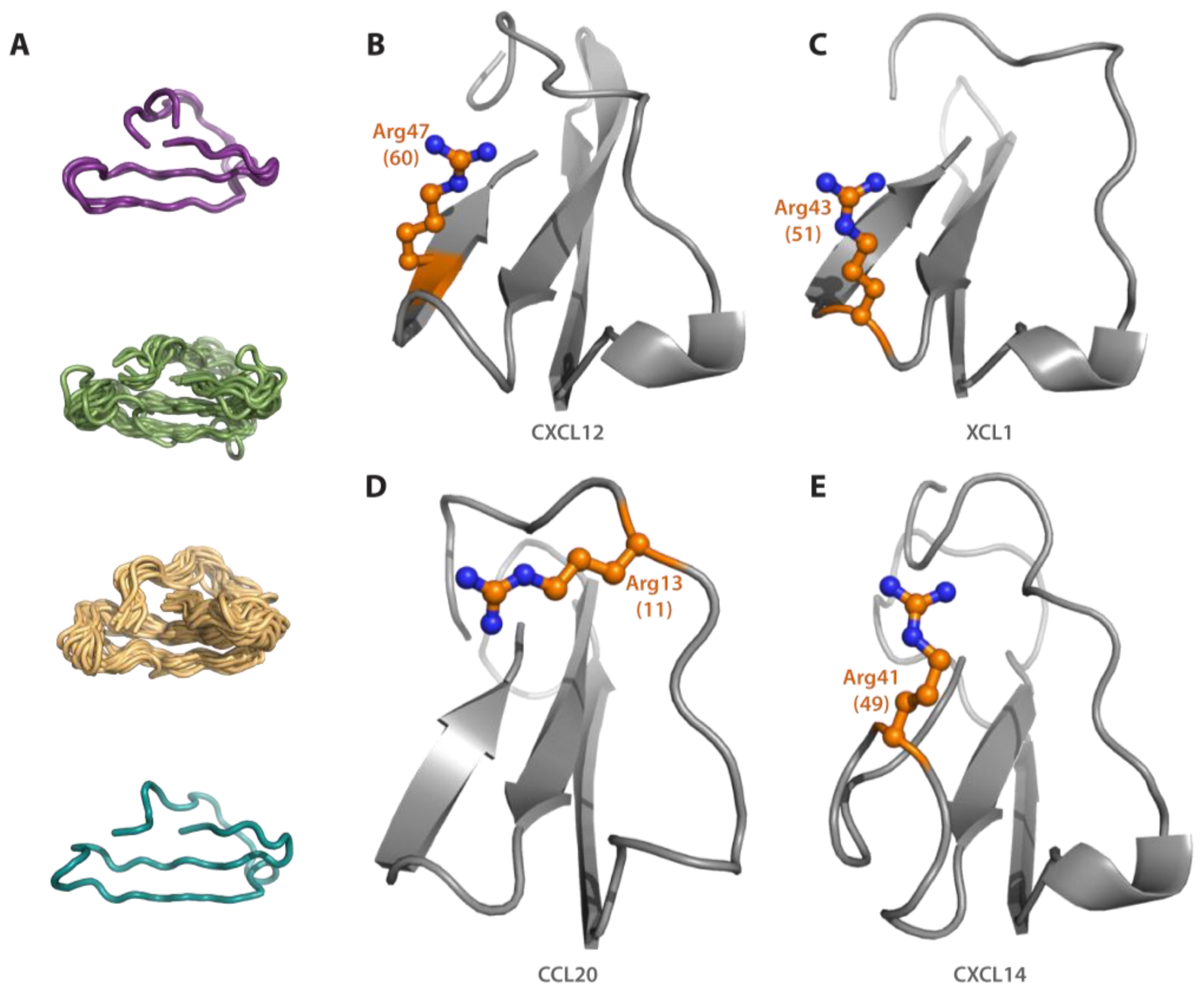
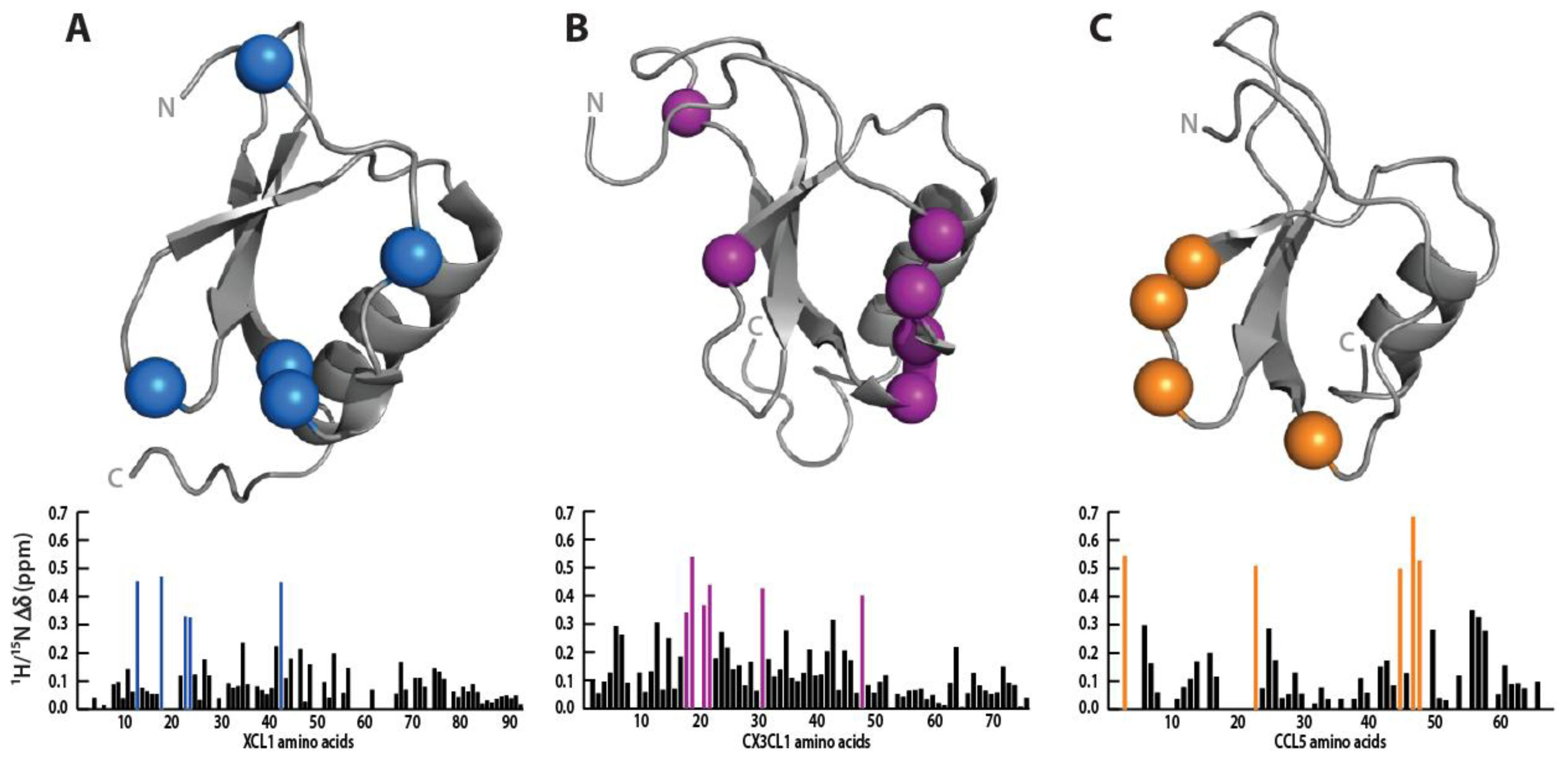
3.5. Sulfotyrosine Probe Highlights Similar Pocket in XCL1, CCL5 and CX3CL1 Chemokines
4. Conclusions
Acknowledgements
- Conflict of InterestThe authors declare no conflict of interest.
References
- Asensio, VC; Campbell, IL. Chemokines in the CNS: Plurifunctional mediators in diverse states. Trends Neurosci 1999, 22, 504–512. [Google Scholar]
- Szekanecz, Z; Kim, J; Koch, AE. Chemokines and chemokine receptors in rheumatoid arthritis. Semin. Immunol 2003, 15, 15–21. [Google Scholar]
- Zernecke, A; Shagdarsuren, E; Weber, C. Chemokines in atherosclerosis: An update. Arterioscler. Thromb. Vasc. Biol 2008, 28, 1897–1908. [Google Scholar]
- Muller, A; Homey, B; Soto, H; Ge, N; Catron, D; Buchanan, ME; McClanahan, T; Murphy, E; Yuan, W; Wagner, SN; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar]
- Crump, MP; Gong, JH; Loetscher, P; Rajarathnam, K; Amara, A; Arenzana-Seisdedos, F; Virelizier, JL; Baggiolini, M; Sykes, BD; Clark-Lewis, I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J 1997, 16, 6996–7007. [Google Scholar]
- Dealwis, C; Fernandez, EJ; Thompson, DA; Simon, RJ; Siani, MA; Lolis, E. Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1alpha, a potent ligand for the HIV-1 “fusin” coreceptor. Proc. Natl. Acad. Sci. USA 1998, 95, 6941–6946. [Google Scholar]
- Baeuerle, PA; Huttner, WB. Tyrosine sulfation is a trans-Golgi-specific protein modification. J. Cell Biol 1987, 105, 2655–2664. [Google Scholar]
- Liu, J; Louie, S; Hsu, W; Yu, KM; Nicholas, HB, Jr; Rosenquist, GL. Tyrosine sulfation is prevalent in human chemokine receptors important in lung disease. Am. J. Respir. Cell Mol. Biol 2008, 38, 738–743. [Google Scholar]
- Monigatti, F; Gasteiger, E; Bairoch, A; Jung, E. The Sulfinator: Predicting tyrosine sulfation sites in protein sequences. Bioinformatics 2002, 18, 769–770. [Google Scholar]
- Farzan, M; Babcock, GJ; Vasilieva, N; Wright, PL; Kiprilov, E; Mirzabekov, T; Choe, H. The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1 alpha association and HIV-1 entry. J. Biol. Chem 2002, 277, 29484–29489. [Google Scholar]
- Farzan, M; Mirzabekov, T; Kolchinsky, P; Wyatt, R; Cayabyab, M; Gerard, NP; Gerard, C; Sodroski, J; Choe, H. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 1999, 96, 667–676. [Google Scholar]
- Fong, AM; Alam, SM; Imai, T; Haribabu, B; Patel, DD. CX3CR1 tyrosine sulfation enhances fractalkine-induced cell adhesion. J. Biol. Chem 2002, 277, 19418–19423. [Google Scholar]
- Preobrazhensky, AA; Dragan, S; Kawano, T; Gavrilin, MA; Gulina, IV; Chakravarty, L; Kolattukudy, PE. Monocyte chemotactic protein-1 receptor CCR2B is a glycoprotein that has tyrosine sulfation in a conserved extracellular N-terminal region. J. Immunol 2000, 165, 5295–5303. [Google Scholar]
- Colvin, RA; Campanella, GS; Manice, LA; Luster, AD. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol. Cell. Biol 2006, 26, 5838–5849. [Google Scholar]
- Stone, MJ; Chuang, S; Hou, X; Shoham, M; Zhu, JZ. Tyrosine sulfation: an increasingly recognised post-translational modification of secreted proteins. New Biotechnol 2009, 25, 299–317. [Google Scholar]
- Veldkamp, CT; Seibert, C; Peterson, FC; De la Cruz, NB; Haugner, JC, III; Basnet, H; Sakmar, TP; Volkman, BF. Structural basis of CXCR 4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal 2008, 1. [Google Scholar] [CrossRef]
- Veldkamp, CT; Ziarek, JJ; Peterson, FC; Chen, Y; Volkman, BF. Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J. Am. Chem. Soc 2010, 132, 7242–7243. [Google Scholar]
- Chenna, R; Sugawara, H; Koike, T; Lopez, R; Gibson, TJ; Higgins, DG; Thompson, JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 2003, 31, 3497–3500. [Google Scholar]
- Crooks, GE; Hon, G; Chandonia, JM; Brenner, SE. WebLogo: A sequence logo generator. Genome Res 2004, 14, 1188–1190. [Google Scholar]
- Berman, HM; Westbrook, J; Feng, Z; Gilliland, G; Bhat, TN; Weissig, H; Shindyalov, IN; Bourne, PE. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar]
- Pieper, U; Eswar, N; Davis, FP; Braberg, H; Madhusudhan, MS; Rossi, A; Marti-Renom, M; Karchin, R; Webb, BM; Eramian, D; et al. MODBASE: A database of annotated comparative protein structure models and associated resources. Nucleic Acids Res 2006, 34, D291–D295. [Google Scholar]
- Pieper, U; Eswar, N; Webb, BM; Eramian, D; Kelly, L; Barkan, DT; Carter, H; Mankoo, P; Karchin, R; Marti-Renom, MA; et al. MODBASE, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res 2009, 37, D347–D354. [Google Scholar]
- Koradi, R; Billeter, M; Wuthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph 1996, 14, 51–55. [Google Scholar]
- Hasegawa, H; Holm, L. Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol 2009, 19, 341–348. [Google Scholar]
- Veldkamp, CT; Peterson, FC; Pelzek, AJ; Volkman, BF. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci 2005, 14, 1071–1081. [Google Scholar]
- Tuinstra, RL; Peterson, FC; Elgin, ES; Pelzek, AJ; Volkman, BF. An engineered second disulfide bond restricts lymphotactin/XCL1 to a chemokine-like conformation with XCR1 agonist activity. Biochemistry 2007, 46, 2564–2573. [Google Scholar]
- Veldkamp, CT; Peterson, FC; Hayes, PL; Mattmiller, JE; Haugner, JC, III; de la Cruz, N; Volkman, BF. On-column refolding of recombinant chemokines for NMR studies biological assays. Protein Expr. Purif 2007, 52, 202–209. [Google Scholar]
- Delaglio, F; Grzesiek, S; Vuister, GW; Zhu, G; Pfeifer, J; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar]
- Chung, CW; Cooke, RM; Proudfoot, AE; Wells, TN. The three-dimensional solution structure of RANTES. Biochemistry 1995, 34, 9307–9314. [Google Scholar]
- Mizoue, LS; Bazan, JF; Johnson, EC; Handel, TM. Solution structure and dynamics of the CX3C chemokine domain of fractalkine and its interaction with an N-terminal fragment of CX3CR1. Biochemistry 1999, 38, 1402–1414. [Google Scholar]
- Keller, R. The Computer Aided Resonance Assignment/Tutorial; CANTINA: Zurich, Switzerland, 2004; pp. 1–73. [Google Scholar]
- Chan, DI; Hunter, HN; Tack, BF; Vogel, HJ. Human macrophage inflammatory protein 3alpha: Protein and peptide nuclear magnetic resonance solution structures, dimerization, dynamics, and anti-infective properties. Antimicrob. Agents Chemother 2008, 52, 883–894. [Google Scholar]
- Veldkamp, CT; Ziarek, JJ; Su, J; Basnet, H; Lennertz, R; Weiner, JJ; Peterson, FC; Baker, JE; Volkman, BF. Monomeric structure of the cardioprotective chemokine SDF-1/CXCL12. Protein Sci 2009, 18, 1359–1369. [Google Scholar]
- Holm, L; Kaariainen, S; Rosenstrom, P; Schenkel, A. Searching protein structure databases with DaliLite v.3. Bioinformatics 2008, 24, 2780–2781. [Google Scholar]
- Holm, L; Kääriäinen, S; Wilton, C; Plewczynski, D. Using Dali for Structural Comparison of Proteins; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2002; pp. 5.5.1–5.5.24. [Google Scholar]
- Veldkamp, CT; Seibert, C; Peterson, FC; Sakmar, TP; Volkman, BF. Recognition of a CXCR4 sulfotyrosine by the chemokine stromal cell-derived factor-1alpha (SDF-1alpha/CXCL12). J. Mol. Biol 2006, 359, 1400–1409. [Google Scholar]
- Mizoue, LS; Sullivan, SK; King, DS; Kledal, TN; Schwartz, TW; Bacon, KB; Handel, TM. Molecular determinants of receptor binding and signaling by the CX3C chemokine fractalkine. J. Biol. Chem 2001, 276, 33906–33914. [Google Scholar]
- Skelton, NJ; Aspiras, F; Ogez, J; Schall, TJ. Proton NMR assignments and solution conformation of RANTES, a chemokine of the C-C type. Biochemistry 1995, 34, 5329–5342. [Google Scholar]
- Duma, L; Haussinger, D; Rogowski, M; Lusso, P; Grzesiek, S. Recognition of RANTES by extracellular parts of the CCR5 receptor. J. Mol. Biol 2007, 365, 1063–1075. [Google Scholar]
- Ye, J; Kohli, LL; Stone, MJ. Characterization of binding between the chemokine eotaxin and peptides derived from the chemokine receptor CCR3. J. Biol. Chem 2000, 275, 27250–27257. [Google Scholar]
- Mayer, MR; Stone, MJ. Identification of receptor binding and activation determinants in the N-terminal and N-loop regions of the CC chemokine eotaxin. J. Biol. Chem 2001, 276, 13911–13916. [Google Scholar]
- Ye, J; Mayer, KL; Mayer, MR; Stone, MJ. NMR solution structure and backbone dynamics of the CC chemokine eotaxin-3. Biochemistry 2001, 40, 7820–7831. [Google Scholar]
- Skelton, NJ; Quan, C; Reilly, D; Lowman, H. Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure 1999, 7, 157–168. [Google Scholar]
- Laurence, JS; Blanpain, C; de Leener, A; Parmentier, M; LiWang, PJ. Importance of basic residues and quaternary structure in the function of MIP-1 beta: CCR5 binding and cell surface sugar interactions. Biochemistry 2001, 40, 4990–4999. [Google Scholar]
- Clubb, RT; Omichinski, JG; Clore, GM; Gronenborn, AM. Mapping the binding surface of interleukin-8 complexed with an N-terminal fragment of the type 1 human interleukin-8 receptor. FEBS Lett 1994, 338, 93–97. [Google Scholar]
- Hemmerich, S; Paavola, C; Bloom, A; Bhakta, S; Freedman, R; Grunberger, D; Krstenansky, J; Lee, S; McCarley, D; Mulkins, M; et al. Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor, CCR2. Biochemistry 1999, 38, 13013–13025. [Google Scholar]
- Zlotnik, A; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar]
- Mayer, KL; Stone, MJ. NMR solution structure and receptor peptide binding of the CC chemokine eotaxin-2. Biochemistry 2000, 39, 8382–8395. [Google Scholar]
- Simpson, LS; Zhu, JZ; Widlanski, TS; Stone, MJ. Regulation of chemokine recognition by site-specific tyrosine sulfation of receptor peptides. Chem. Biol 2009, 16, 153–161. [Google Scholar]
- Simpson, LS; Widlanski, TS. A comprehensive approach to the synthesis of sulfate esters. J. Am. Chem. Soc 2006, 128, 1605–1610. [Google Scholar]
- Seibert, C; Veldkamp, CT; Peterson, FC; Chait, BT; Volkman, BF; Sakmar, TP. Sequential tyrosine sulfation of CXCR4 by tyrosylprotein sulfotransferases. Biochemistry 2008, 47, 11251–11262. [Google Scholar]
- Ziarek, JJ; Peterson, FC; Lytle, BL; Volkman, BF. Binding site identification and structure determination of protein-ligand complexes by NMR a semiautomated approach. Methods Enzymol 2011, 493, 241–275. [Google Scholar]
- Wieting, J; Volkman, BF. Personal communication, Medical College of Wisconsin: Milwaukee, WI, 2011.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a)
| ||||
|---|---|---|---|---|
| Chemokine | PDB ID | Dali Z-score | Cα RMSD (Å) | Sequence Identity (%) |
| XCL Subfamily | 2HDM | |||
| XCL 1 | ||||
| CCL Subfamily | ||||
| CCL1 | IEL0 | 3.6 | 2.3 | 30 |
| CCL2 | 1DOM | 5.6 | 1.3 | 25 |
| CCL3 | QB53 | 5.3 | 1.6 | 50 |
| CCL4 | 1HUM | 5.4 | 1.7 | 55 |
| CCL5 | 1HRJ | |||
| CCL7 | 1BO0 | 5.1 | 1.5 | 28 |
| CCL8 | 1ESR | 5.8 | 1.5 | 33 |
| CCL11 | 1EOT | 5.1 | 1.5 | 30 |
| CCL13 | 2RA4 | 5.8 | 1.5 | 28 |
| CCL14 | 2Q8T | 6.1 | 1.3 | 40 |
| CCL15 | 2HCC | 5.3 | 1.5 | 50 |
| CCL17 | 1NR4 | 5.6 | 1.5 | 38 |
| CCL20 | 2JYO | 5.7 | 1.4 | 30 |
| CCL23 | 1G91 | 5.3 | 1.5 | 40 |
| CCL24 | 1EIG | 5.1 | 1.6 | 28 |
| CCL26 | 1G2S | 5.8 | 1.5 | 43 |
| CCL27 | 2KUM | 4.0 | 2.0 | 21 |
| CXCL subfamily | ||||
| CXCL1 | 1MSG | 2.1 | 2.8 | 14 |
| CXCL2 | 1QNK | 5.2 | 2.0 | 20 |
| CXCL4 | 1F9A | 4.8 | 1.9 | 26 |
| CXCL7 | 1NAP | 5.2 | 1.8 | 23 |
| CXCL8 | 1ILQ | 4.0 | 2.0 | 21 |
| CXCL10 | 1LV9 | 3.7 | 2.6 | 15 |
| CXCL11 | 1RJT | 1.7 | 2.6 | 9 |
| CXCL12 | 2KEE | |||
| CXCL14 | 2HDL | 4.6 | 2.6 | 20 |
| CX3CL subfamily | ||||
| CX3CL1 | 1B2T | 4.0 | 2.3 | 18 |
| (b)
| ||||||
|---|---|---|---|---|---|---|
| Chemokine | Mod Base ID | Template PDB | MPQS Score | Dali Z-score | Cα RMSD (Å) | Sequence Identity (%) |
| XCL subfamily | ||||||
| XCL2 | Q9UBD3 | 1J8I (XCL1) | 1.85 | 5.1 | 1.2 | 97 |
| CCL subfamily | ||||||
| CCL16 | O15467 | 2Q8R (CCL4) | 1.16 | 5.6 | 1.1 | 41 |
| CCL18 | P55774 | 1ZXT (viral) | 1.40 | 5.6 | 1.1 | 35 |
| CCL19 | Q99731 | 1ZXT (viral) | 1.21 | 5.4 | 1.5 | 33 |
| CCL21 | O00585 | 1ESR (CCL8) | 1.11 | 4.8 | 1.5 | 33 |
| CCL22 | O00626 | 1ZXT (viral) | 1.25 | 5.6 | 1.5 | 33 |
| CXCL subfamily | ||||||
| CXCL3 | P19876 | 1QNK (CXCL2) | 1.70 | 4.7 | 2.0 | 18 |
| CXCL5 | P42830 | 1TVX (CXCL7) | 1.27 | 5.0 | 1.9 | 23 |
| CXCL6 | P27784 | 1TVX (CXCL7) | 1.22 | 4.4 | 1.8 | 26 |
| CXCL9 | Q07325 | 1GNK(CXCL2) | 1.18 | 4.8 | 1.8 | 28 |
| CXCL13 | Q43927 | 3IL8(CXCL8) | 1.26 | 4.3 | 1.9 | 26 |
© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ziarek, J.J.; Heroux, M.S.; Veldkamp, C.T.; Peterson, F.C.; Volkman, B.F. Sulfotyrosine Recognition as Marker for Druggable Sites in the Extracellular Space. Int. J. Mol. Sci. 2011, 12, 3740-3756. https://doi.org/10.3390/ijms12063740
Ziarek JJ, Heroux MS, Veldkamp CT, Peterson FC, Volkman BF. Sulfotyrosine Recognition as Marker for Druggable Sites in the Extracellular Space. International Journal of Molecular Sciences. 2011; 12(6):3740-3756. https://doi.org/10.3390/ijms12063740
Chicago/Turabian StyleZiarek, Joshua J., Maxime S. Heroux, Christopher T. Veldkamp, Francis C. Peterson, and Brian F. Volkman. 2011. "Sulfotyrosine Recognition as Marker for Druggable Sites in the Extracellular Space" International Journal of Molecular Sciences 12, no. 6: 3740-3756. https://doi.org/10.3390/ijms12063740