Acid-Denatured Green Fluorescent Protein (GFP) as Model Substrate to Study the Chaperone Activity of Protein Disulfide Isomerase



Abstract
:
1. Introduction
2. Results and Discussion
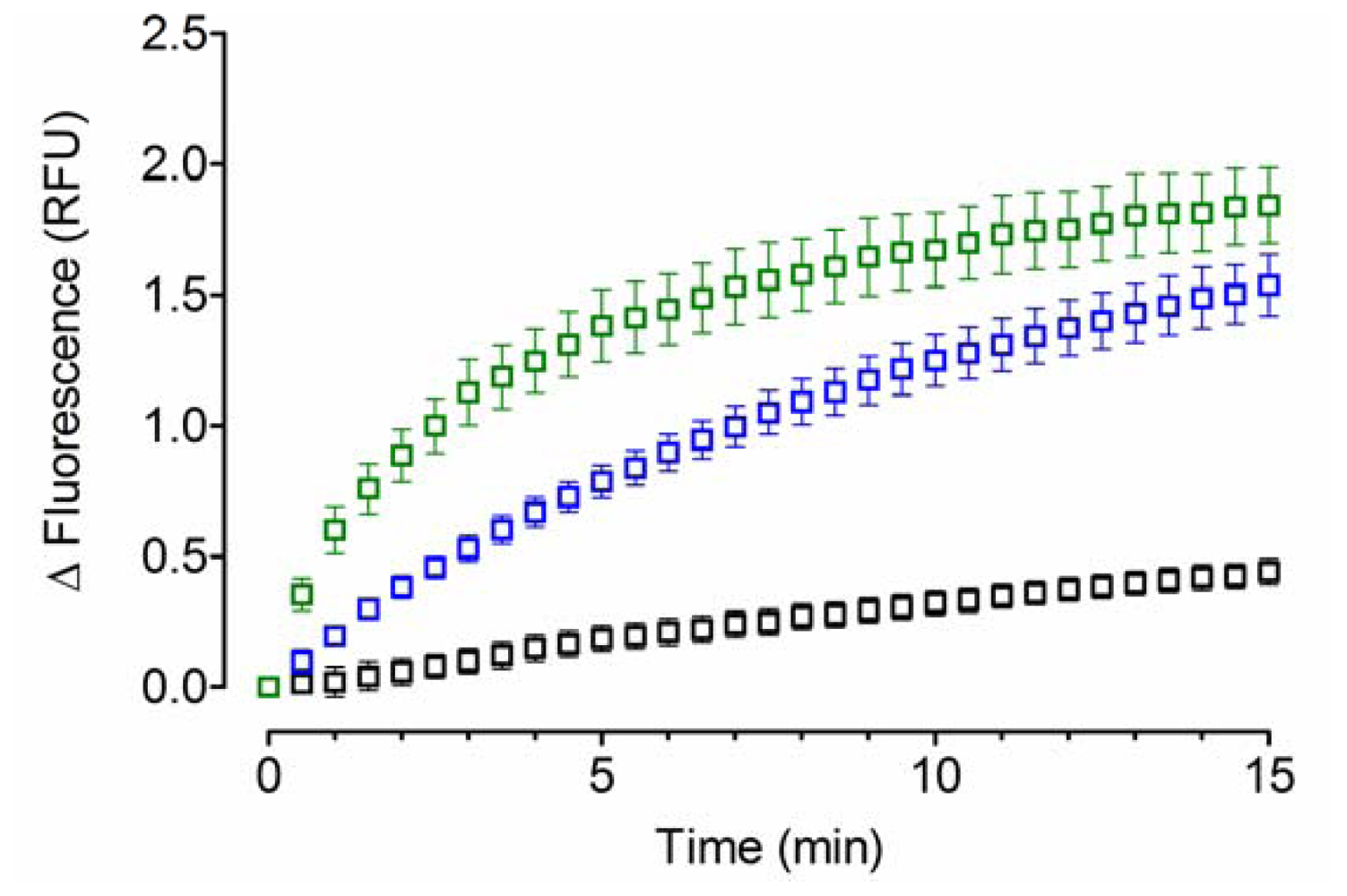
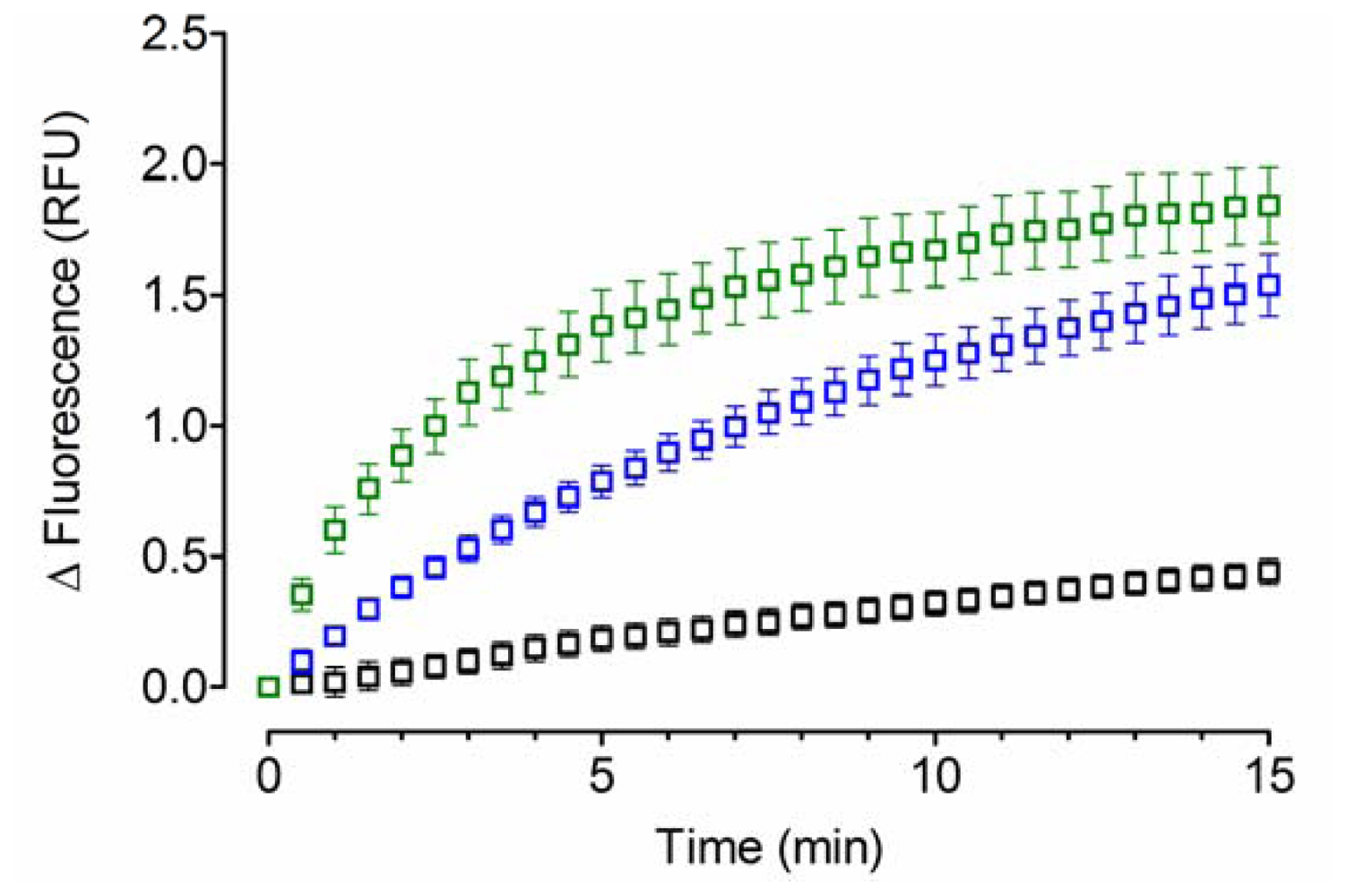
2.1. Acid-Denatured GFP as Model Substrate
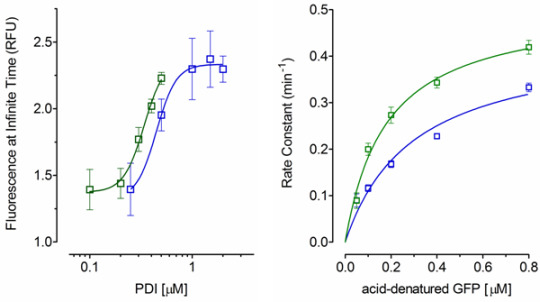
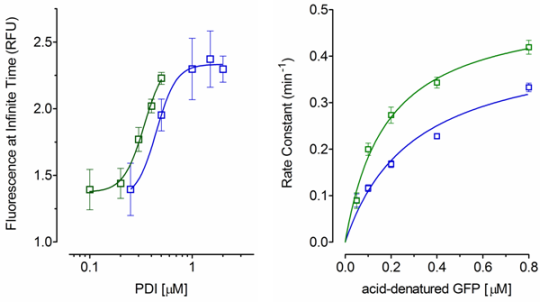
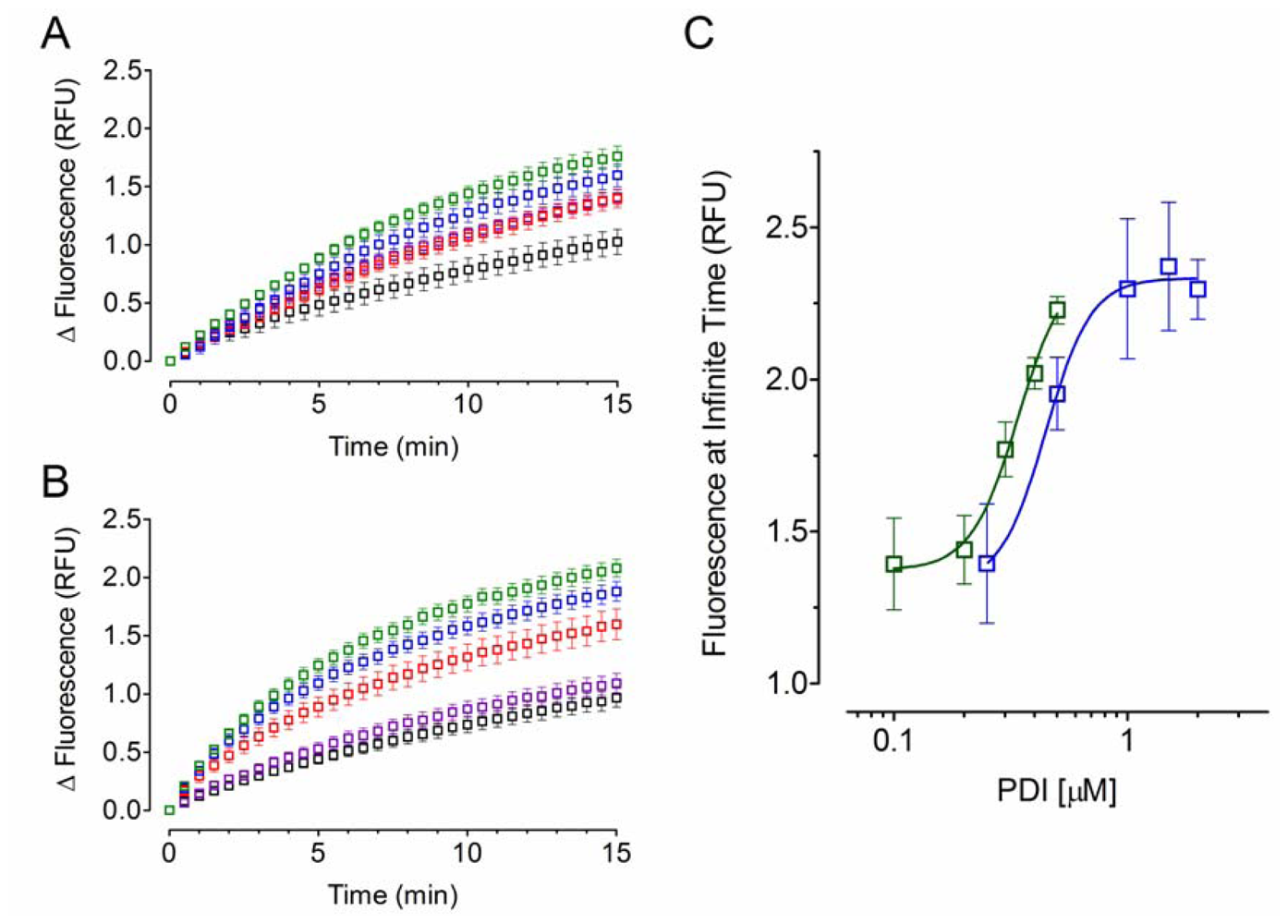
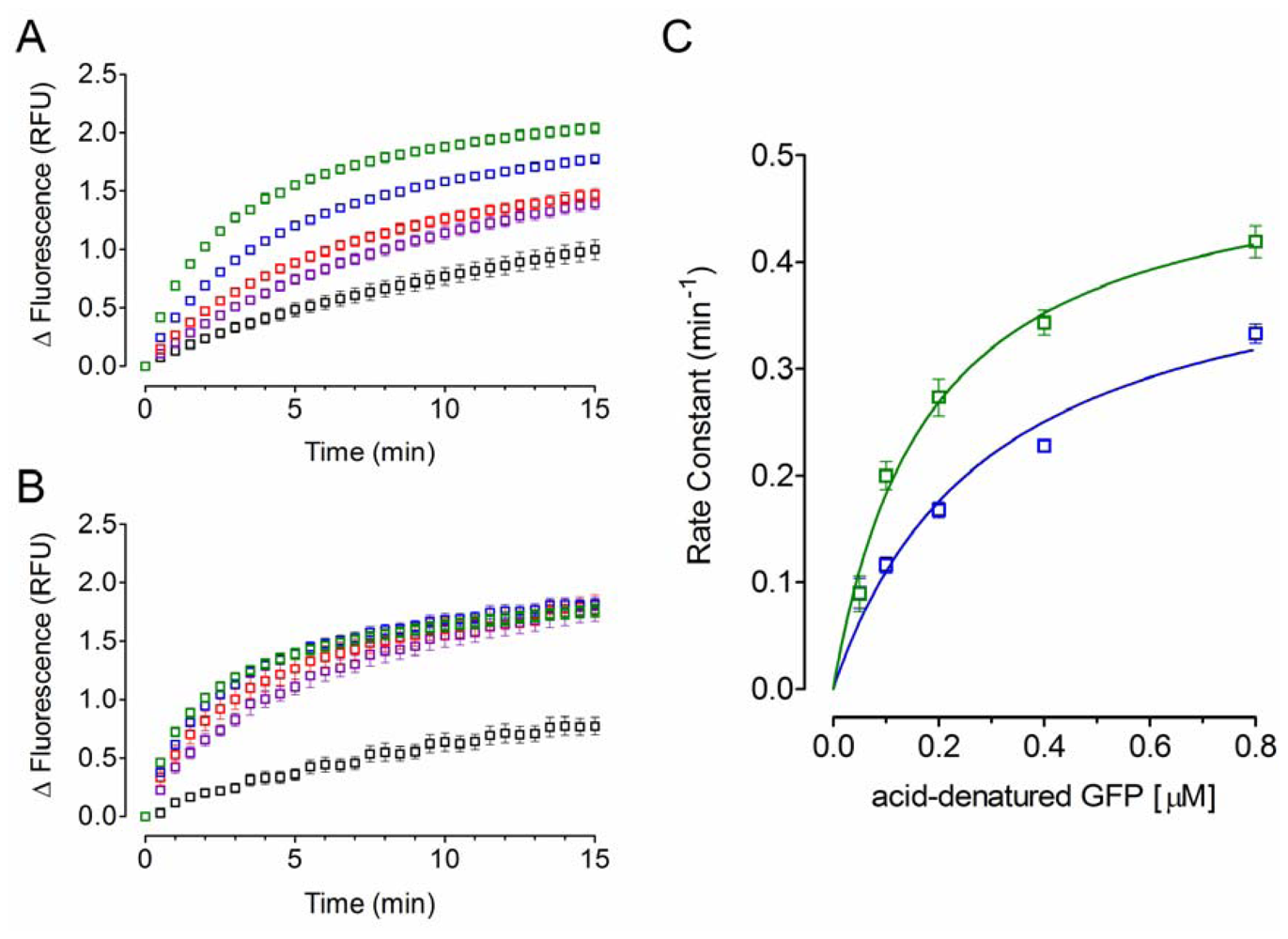
2.2. Effect of PDI Concentration
2.3. Effect of Acid-Denatured GFP Concentration
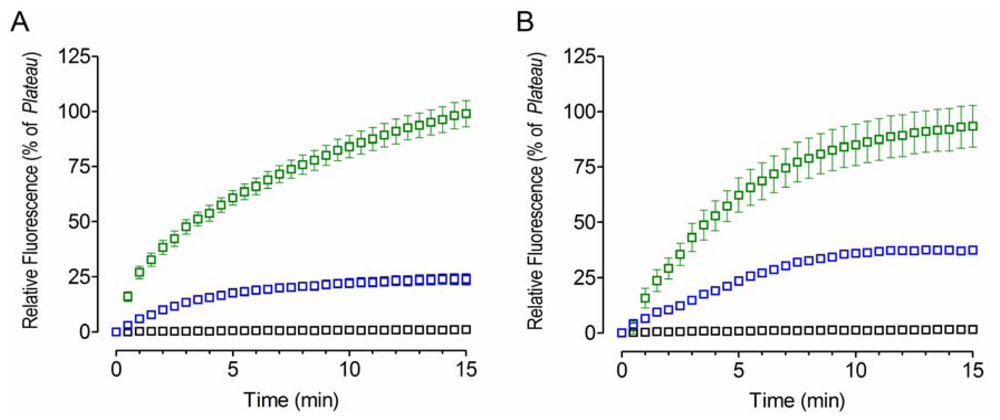
2.4. Bacitracin Inhibits the Chaperone Activity of PDI
3. Experimental Section
3.1. Vectors, Bacterial Strains, Enzymes and Chemicals
3.2. Construction of Recombinant Plasmids
3.3. Expression and Purification of GFP, YPDI and HuPDI
GFP, YPDI and HuPDI expression
GFP purification
YPDI and HuPDI purification
3.4. Acid-Denaturation of GFP
3.5. Refolding of GFP by PDI Proteins
3.6. Inhibition of the PDI Chaperone Activity by Bacitracin
3.7. Statistical and Data Analysis
4. Conclusions
Acknowledgements
References
- Shimomura, O; Johnson, FH; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell. Comp. Physiol 1962, 59, 223–239. [Google Scholar]
- Heim, R; Prasher, DC; Tsien, RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar]
- Bizzarri, R; Serresi, M; Luin, S; Beltram, F. Green fluorescent protein based pH indicators for in vivo use: A review. Anal. Bioanal. Chem 2009, 393, 1107–1122. [Google Scholar]
- Morris, MC. Fluorescent biosensors of intracellular targets from genetically encoded reporters to modular polypeptide probes. Cell. Biochem. Biophys 2010, 56, 19–37. [Google Scholar]
- Ward, WW; Bokman, SH. Reversible denaturation of Aequorea green-fluorescent protein: physical separation and characterization of the renatured protein. Biochemistry 1982, 21, 4535–4540. [Google Scholar]
- Penna, TC; Ishii, M; Junior, AP; Cholewa, O. Thermal stability of recombinant green fluorescent protein (GFPuv) at various pH values. Appl. Biochem. Biotechnol 2004, 114, 469–483. [Google Scholar]
- Nagy, A; Malnasi-Csizmadia, A; Somogyi, B; Lorinczy, D. Thermal stability of chemically denatured green fluorescent protein (GFP)—A preliminary study. Thermochim. Acta 2004, 410, 161–163. [Google Scholar]
- Alkaabi, KM; Yafea, A; Ashraf, SS. Effect of pH on thermal- and chemical-induced denaturation of GFP. Appl. Biochem. Biotechnol 2005, 126, 149–156. [Google Scholar]
- Enoki, S; Saeki, K; Maki, K; Kuwajima, K. Acid denaturation and refolding of green fluorescent protein. Biochemistry 2004, 43, 14238–14248. [Google Scholar]
- Makino, Y; Amada, K; Taguchi, H; Yoshida, M. Chaperonin-mediated folding of green fluorescent protein. J. Biol. Chem 1997, 272, 12468–12474. [Google Scholar]
- Yoshida, T; Kawaguchi, R; Taguchi, H; Yoshida, M; Yasunaga, T; Wakabayashi, T; Yohda, M; Maruyama, T. Archaeal group II chaperonin mediates protein folding in the cis-cavity without a detachable GroES-like co-chaperonin. J. Mol. Biol 2002, 315, 73–85. [Google Scholar]
- Okochi, M; Matsuzaki, H; Nomura, T; Ishii, N; Yohda, M. Molecular characterization of the group II chaperonin from the hyperthermophilic archaeum Pyrococcus horikoshii OT3. Extremophiles 2005, 9, 127–134. [Google Scholar]
- Wang, L; Hu, ZJ; Luo, YM; Huo, YW; Ma, Q; He, YZ; Zhang, YY; Sun, F; Dong, ZY. Distinct symmetry and limited peptide refolding activity of the thermosomes from the acidothermophilic archaea Acidianus tengchongensis S5(T). Biochem. Biophys. Res. Commun 2010, 393, 228–234. [Google Scholar]
- Shi, R; Pan, Q; Guan, Y; Hua, Z; Huang, Y; Zhao, M; Li, Y. Imidazole as a catalyst for in vitro refolding of enhanced green fluorescent protein. Arch. Biochem. Biophys 2007, 459, 122–128. [Google Scholar]
- Asayama, W; Sawada, S; Taguchi, H; Akiyoshi, K. Comparison of refolding activities between nanogel artificial chaperone and GroEL systems. Int. J. Biol. Macromol 2008, 42, 241–246. [Google Scholar]
- Wang, C; Chen, S; Wang, X; Wang, L; Wallis, AK; Freedman, RB; Wang, CC. Plasticity of human protein disulfide isomerase: evidence for mobility around the X-linker region and its functional significance. J. Biol. Chem 2010, 285, 26788–26797. [Google Scholar]
- Wilkinson, B; Gilbert, HF. Protein disulfide isomerase. Biochim. Biophys. Acta 2004, 1699, 35–44. [Google Scholar]
- Katiyar, S; Till, EA; Lennarz, WJ. Studies on the function of yeast protein disulfide isomerase in renaturation of proteins. Biochim. Biophys. Acta 2001, 1548, 47–56. [Google Scholar]
- Hatahet, F; Ruddock, LW. Substrate recognition by the protein disulfide isomerases. FEBS J 2007, 274, 5223–5234. [Google Scholar]
- Kozlov, G; Määttänen, P; Thomas, DY; Gehring, K. A structural overview of the PDI family of proteins. FEBS J 2010, 277, 3924–3936. [Google Scholar]
- Fu, XM; Zhu, BT. Human pancreas-specific protein disulfide-isomerase (PDIp) can function as a chaperone independently of its enzymatic activity by forming stable complexes with denatured substrate proteins. Biochem. J 2010, 429, 157–169. [Google Scholar]
- Spinozzi, F; Mariani, P; Rustichelli, F; Amenitsch, H; Bennardini, F; Mura, GM; Coi, A; Ganadu, ML. Temperature dependence of chaperone-like activity and oligomeric state of alphaB-crystallin. Biochim. Biophys. Acta 2006, 1764, 677–687. [Google Scholar]
- Pirneskoski, A; Klappa, P; Lobell, M; Williamson, RA; Byrne, L; Alanen, HI; Salo, KE; Kivirikko, KI; Freedman, RB; Ruddock, LW. Molecular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein disulfide isomerase. J. Biol. Chem 2004, 279, 10374–10381. [Google Scholar]
- Nguyen, VD; Wallis, K; Howard, MJ; Haapalainen, AM; Salo, KE; Saaranen, MJ; Sidhu, A; Wierenga, RK; Freedman, RB; Ruddock, LW; Williamson, RA. Alternative conformations of the x region of human protein disulphide-isomerase modulate exposure of the substrate binding b’ domain. J. Mol. Biol 2008, 383, 1144–1155. [Google Scholar]
- Byrne, LJ; Sidhu, A; Wallis, AK; Ruddock, LW; Freedman, RB; Howard, MJ; Williamson, RA. Mapping of the ligand-binding site on the b’ domain of human PDI: Interaction with peptide ligands and the x-linker region. Biochem. J 2009, 423, 209–217. [Google Scholar]
- Mandel, R; Ryser, HJ; Ghani, F; Wu, M; Peak, D. Inhibition of a reductive function of the plasma membrane by bacitracin and antibodies against protein disulfide-isomerase. Proc. Natl. Acad. Sci. USA 1993, 90, 4112–4116. [Google Scholar]
- Versteeg, HH; Ruf, W. Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J. Biol. Chem 2007, 282, 25416–25424. [Google Scholar]
- Tachibana, C; Stevens, TH. The yeast EUG1 gene encodes an endoplasmic reticulum protein that is functionally related to protein disulfide isomerase. Mol. Cell. Biol 1992, 12, 4601–4611. [Google Scholar]
- Pace, CN; Vajdos, F; Fee, L; Grimsley, G; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci 1995, 4, 2411–2423. [Google Scholar]
- Tanaka, S; Uehara, T; Nomura, Y. Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J. Biol. Chem 2000, 275, 10388–10293. [Google Scholar]
- Rao, RV; Ellerby, HM; Bredesen, DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ 2004, 11, 372–380. [Google Scholar]
- Barbouche, R; Miquelis, R; Jones, IM; Fenouillet, E. Protein-disulfide isomerase-mediated reduction of two disulfide bonds of HIV envelope glycoprotein 120 occurs post-CXCR4 binding and is required for fusion. J. Biol. Chem 2003, 278, 3131–3136. [Google Scholar]
- Abromaitis, S; Stephens, RS. Attachment and entry of Chlamydia have distinct requirements for host protein disulfide isomerase. PLoS Pathog 2009, 5, e1000357. [Google Scholar]
- Santos, CX; Stolf, BS; Takemoto, PV; Amanso, AM; Lopes, LR; Souza, EB; Goto, H; Laurindo, FR. Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of Leishmania chagasi promastigotes by macrophages. J. Leukoc. Biol 2009, 86, 989–998. [Google Scholar]
- Corazzari, M; Lovat, PE; Armstrong, JL; Fimia, GM; Hill, DS; Birch-Machin, M; Redfern, CP; Piacentini, M. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: The role of stress proteins ERdj5 and ERp57. Br. J. Cancer 2007, 96, 1062–1071. [Google Scholar]
- Lovat, PE; Corazzari, M; Armstrong, JL; Martin, S; Pagliarini, V; Hill, D; Brown, AM; Piacentini, M; Birch-Machin, MA; Redfern, CP. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res 2008, 68, 5363–5369. [Google Scholar]
- Gallina, A; Hanley, TM; Mandel, R; Trahey, M; Broder, CC; Viglianti, GA; Ryser, HJ. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J. Biol. Chem 2002, 277, 50579–50588. [Google Scholar]
- Smith, AM; Chan, J; Oksenberg, D; Urfer, R; Wexler, DS; Ow, A; Gao, L; McAlorum, A; Huang, SG. A high-throughput turbidometric assay for screening inhibitors of protein disulfide isomerase. J. Biomol. Screen 2004, 9, 614–620. [Google Scholar]
- Khalaf, NB; De Muylder, G; Ratnam, J; Ang, KK-H; Arkin, M; McKerrow, J; Chenik, M. A high-throughput turbidometric assay for screening inhibitors of Leishmania major protein disulfide isomerase. J. Biomol. Screen 2011, 16, 545–551. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spontaneous | YPDI | HuPDI | |
|---|---|---|---|
| k ( min−1) | 0.037 ± 0.021 | 0.107 ± 0.012 | 0.311 ± 0.027 |
| Plateau (RFU) | 1.04 ± 0.48 | 1.91 ± 0.12 | 1.78 ± 0.04 |
| EC50 (μM) | n.d. | 0.45 ± 0.09 | 0.34 ± 0.04 |
| Hill coefficient | n.d. | 0.89 ± 0.11 | 1.06 ± 0.08 |
| Kd (μM) | n.d. | 0.30 ± 0.09 | 0.18 ± 0.03 |
| Primer | Sequence | Endonuclease |
|---|---|---|
| ScPDIF | acactcggatccCAACAAGAGGCTGTGGCC | BamHI |
| ScPDIR | acactcctgcagTTACAATTCATCGTGAATGGC | PstI |
| BamPDIH | cgggatccGACGCCCCCGAGGAGGAGGAC | BamHI |
| HsPDIR2 | gcccaagcttACAGTTCATCTTTCACAGCTTTCTG | HindIII |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mares, R.E.; Meléndez-López, S.G.; Ramos, M.A. Acid-Denatured Green Fluorescent Protein (GFP) as Model Substrate to Study the Chaperone Activity of Protein Disulfide Isomerase. Int. J. Mol. Sci. 2011, 12, 4625-4636. https://doi.org/10.3390/ijms12074625
Mares RE, Meléndez-López SG, Ramos MA. Acid-Denatured Green Fluorescent Protein (GFP) as Model Substrate to Study the Chaperone Activity of Protein Disulfide Isomerase. International Journal of Molecular Sciences. 2011; 12(7):4625-4636. https://doi.org/10.3390/ijms12074625
Chicago/Turabian StyleMares, Rosa E., Samuel G. Meléndez-López, and Marco A. Ramos. 2011. "Acid-Denatured Green Fluorescent Protein (GFP) as Model Substrate to Study the Chaperone Activity of Protein Disulfide Isomerase" International Journal of Molecular Sciences 12, no. 7: 4625-4636. https://doi.org/10.3390/ijms12074625