Microcystin-LR Induces Apoptosis via NF-κB /iNOS Pathway in INS-1 Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Reagents
2.2. Cell Culture
2.3. MTT Assay
2.4. Hoechst/PI Staining
2.5. Flow Cytometry
2.6. Immunofluorescence Microscopy
2.7. Transient Transfection and Luciferase Reporter Assay
2.8. Real-Time RT-PCR Assay
2.9. Western Blot
2.10. NO Assay
2.11. Statistical Analysis
3. Results
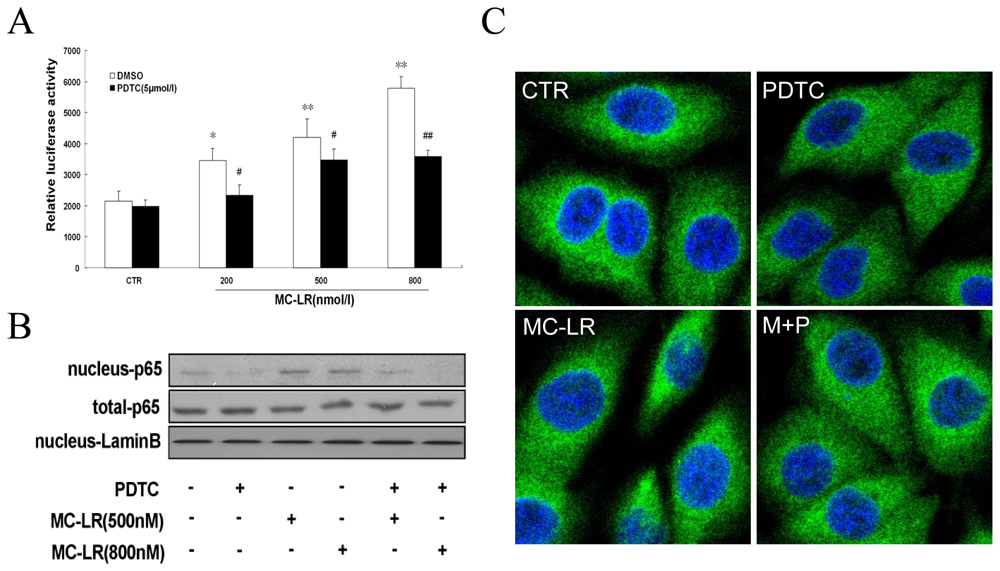
3.1. MC-LR Selectively Promotes Activation of NF-κB in INS-1 cells
3.2. MC-LR Up-Regulates iNOS Expression and Stimulates NO Formation in INS-1 Cells
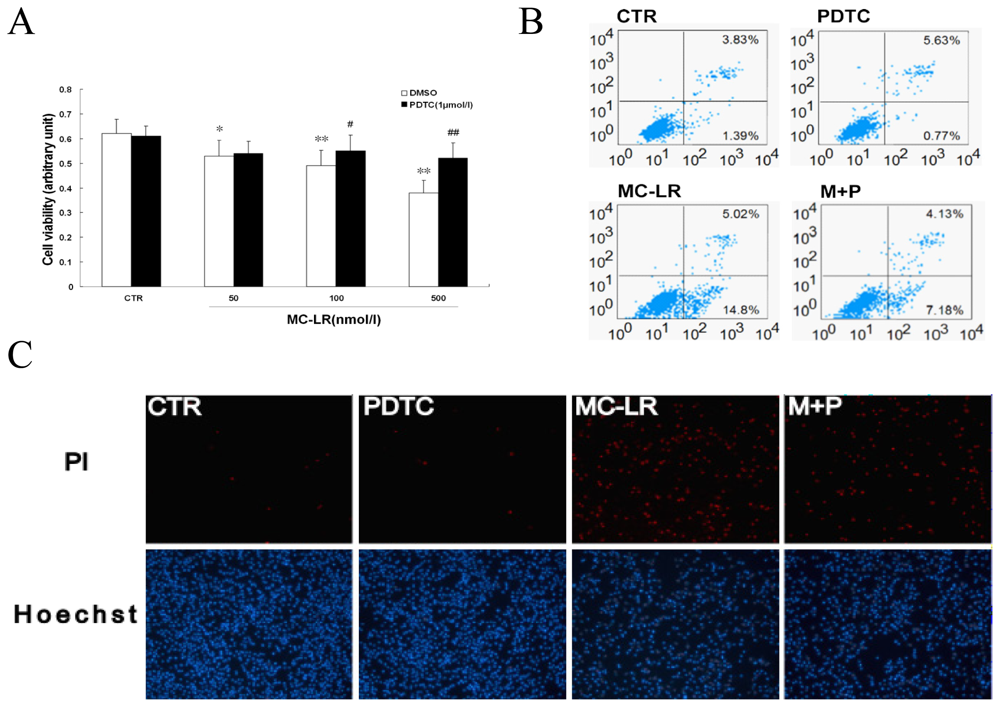
3.3. Chronic Treatment with MC-LR Induces Apoptosis in INS-1 Cells
4. Discussion
Acknowledgements
References
- Honkanen, RE; Zwiller, J; Moore, RE; Daily, SL; Khatra, BS; Dukelow, M; Boynton, AL. Characterization of microcystin-LR, a potent inhibitor of type 1 and type 2A protein phosphatases. J. Biol. Chem 1990, 265, 19401–19404. [Google Scholar]
- Chen, T; Shen, P; Zhang, J; Hua, Z. Effects of microcystin-LR on patterns of iNOS and cytokine mRNA expression in macrophages in vitro. Environ. Toxicol 2005, 20, 85–91. [Google Scholar]
- Weng, D; Lu, Y; Wei, Y; Liu, Y; Shen, P. The role of ROS in microcystin-LR-induced hepatocyte apoptosis and liver injury in mice. Toxicology 2007, 232, 15–23. [Google Scholar]
- Lankoff, A; Krzowski, L; Glab, J; Banasik, A; Lisowska, H; Kuszewski, T; Gozdz, S; Wojcik, A. DNA damage and repair in human peripheral blood lymphocytes following treatment with microcystin-LR. Mutat. Res 2004, 559, 131–142. [Google Scholar]
- McDermott, CM; Nho, CW; Howard, W; Holton, B. The cyanobacterial toxin, microcystin-LR, can induce apoptosis in a variety of cell types. Toxicon 1998, 36, 1981–1996. [Google Scholar]
- Brzuzan, P; Wozny, M; Ciesielski, S; Luczynski, MK; Gora, M; Kuzminski, H; Dobosz, S. Microcystin-LR induced apoptosis and mRNA expression of p53 and cdkn1a in liver of whitefish (Coregonus lavaretus L.). Toxicon 2009, 54, 170–183. [Google Scholar]
- Botha, N; Gehringer, MM; Downing, TG; van de Venter, M; Shephard, EG. The role of microcystin-LR in the induction of apoptosis and oxidative stress in CaCo2 cells. Toxicon 2004, 43, 85–92. [Google Scholar]
- Feng, G; Abdalla, M; Li, Y; Bai, Y. NF-kappaB mediates the induction of Fas receptor and Fas ligand by microcystin-LR in HepG2 cells. Mol. Cell. Biochem 2011, 352, 209–219. [Google Scholar]
- Mikhailov, A; Harmala-Brasken, AS; Hellman, J; Meriluoto, J; Eriksson, JE. Identification of ATP-synthase as a novel intracellular target for microcystin-LR. Chem. Biol. Interact 2003, 142, 223–237. [Google Scholar]
- Tak, PP; Firestein, GS. NF-kappaB: A key role in inflammatory diseases. J. Clin. Invest 2001, 107, 7–11. [Google Scholar]
- Czyz, M. Specificity and selectivity of the NFkappaB response. Postepy. Biochem 2005, 51, 60–68. [Google Scholar]
- Velez-Pardo, C; Morales, AT; Del-Rio, MJ; Olivera-Angel, M. Endogenously generated hydrogen peroxide induces apoptosis via mitochondrial damage independent of NF-kappaB and p53 activation in bovine embryos. Theriogenology 2007, 67, 1285–1296. [Google Scholar]
- Darville, MI; Eizirik, DL. Cytokine induction of Fas gene expression in insulin-producing cells requires the transcription factors NF-kappaB and C/EBP. Diabetes 2001, 50, 1741–1748. [Google Scholar]
- Wurzer, WJ; Ehrhardt, C; Pleschka, S; Berberich-Siebelt, F; Wolff, T; Walczak, H; Planz, O; Ludwig, S. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J. Biol. Chem 2004, 279, 30931–30937. [Google Scholar]
- Dvorak, Z; Pavek, P. Comment on “The role of redox-sensitive transcription factors NF-kB and AP-1 in the modulation of the Cyp1A1 gene by mercury, lead, and copper”. Free Radic. Biol. Med 2008, 45, 939–940. [Google Scholar]
- Kong, G; Kim, E; Kim, W; Lee, Y; Lee, J; Paik, S; Rhee, J; Choi, K; Lee, K. Inducible nitric oxide synthase (iNOS) immunoreactivity and its relationship to cell proliferation, apoptosis, angiogenesis, clinicopathologic characteristics, and patient survival in pancreatic cancer. Int. J. Gastrointest Cancer 2001, 29, 133–140. [Google Scholar]
- Wink, DA; Mitchell, JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med 1998, 25, 434–456. [Google Scholar]
- Siamwala, JH; Majumder, S; Tamilarasan, KP; Muley, A; Reddy, SH; Kolluru, GK; Sinha, S; Chatterjee, S. Simulated microgravity promotes nitric oxide-supported angiogenesis via the iNOS-cGMP-PKG pathway in macrovascular endothelial cells. FEBS Lett 2010, 584, 3415–3423. [Google Scholar]
- Gookin, JL; Chiang, S; Allen, J; Armstrong, MU; Stauffer, SH; Finnegan, C; Murtaugh, MP. NF-kappaB-mediated expression of iNOS promotes epithelial defense against infection by cryptosporidium parvum in neonatal piglets. Am. J. Physiol. Gastrointest Liver Physiol 2006, 290, G164–G174. [Google Scholar]
- Tian, B; Liu, J; Bitterman, PB; Bache, RJ. Mechanisms of cytokine induced NO-mediated cardiac fibroblast apoptosis. Am. J. Physiol. Heart Circ. Physiol 2002, 283, H1958–H1967. [Google Scholar]
- Corbett, JA; Kwon, G; Misko, TP; Rodi, CP; McDaniel, ML. Tyrosine kinase involvement in IL-1 beta-induced expression of iNOS by beta-cells purified from islets of Langerhans. Am. J. Physiol 1994, 267, C48–C54. [Google Scholar]
- Medeiros, R; Prediger, RD; Passos, GF; Pandolfo, P; Duarte, FS; Franco, JL; Dafre, AL; Giunta, G; Figueiredo, CP; Takahashi, RN; et al. Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: Relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J. Neurosci 2007, 27, 5394–5404. [Google Scholar]
- Gurgul, E; Lortz, S; Tiedge, M; Jorns, A; Lenzen, S. Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 2004, 53, 2271–2280. [Google Scholar]
- Li, F; Mahato, RI. iNOS gene silencing prevents inflammatory cytokine-induced beta-cell apoptosis. Mol. Pharm 2008, 5, 407–417. [Google Scholar]
- Han, X; Sun, Y; Scott, S; Bleich, D. Tissue inhibitor of metalloproteinase-1 prevents cytokine-mediated dysfunction and cytotoxicity in pancreatic islets and beta-cells. Diabetes 2001, 50, 1047–1055. [Google Scholar]
- Meng, ZX; Nie, J; Ling, JJ; Sun, JX; Zhu, YX; Gao, L; Lv, JH; Zhu, DY; Sun, YJ; Han, X. Activation of liver X receptors inhibits pancreatic islet beta cell proliferation through cell cycle arrest. Diabetologia 2009, 52, 125–135. [Google Scholar]
- Song, JD; Lee, SK; Kim, KM; Kim, JW; Kim, JM; Yoo, YH; Park, YC. Redox factor-1 mediates NF-kappaB nuclear translocation for LPS-induced iNOS expression in murine macrophage cell line RAW 264.7. Immunology 2008, 124, 58–67. [Google Scholar]
- Angel, P; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar]
- Tseng, CP; Ely, BD; Pong, RC; Wang, Z; Zhou, J; Hsieh, JT. The role of DOC-2/DAB2 protein phosphorylation in the inhibition of AP-1 activity. An underlying mechanism of its tumor-suppressive function in prostate cancer. J. Biol. Chem 1999, 274, 31981–31986. [Google Scholar]
- Ozaki, T; Habara, K; Matsui, K; Kaibori, M; Kwon, AH; Ito, S; Nishizawa, M; Okumura, T. Dexamethasone inhibits the induction of iNOS gene expression through destabilization of its mRNA in proinflammatory cytokine-stimulated hepatocytes. Shock 2010, 33, 64–69. [Google Scholar]
- Cook, T; Wang, Z; Alber, S; Liu, K; Watkins, SC; Vodovotz, Y; Billiar, TR; Blumberg, D. Nitric oxide and ionizing radiation synergistically promote apoptosis and growth inhibition of cancer by activating p53. Cancer Res 2004, 64, 8015–8021. [Google Scholar]
- Qiu, LQ; Sinniah, R; Hsu, SI. Coupled induction of iNOS and p53 upregulation in renal resident cells may be linked with apoptotic activity in the pathogenesis of progressive IgA nephropathy. J. Am. Soc. Nephrol 2004, 15, 2066–2078. [Google Scholar]
- Ding, WX; Shen, HM; Ong, CN. Critical role of reactive oxygen species and mitochondrial permeability transition in microcystin-induced rapid apoptosis in rat hepatocytes. Hepatology 2000, 32, 547–555. [Google Scholar]
- Wei, Y; Weng, D; Li, F; Zou, X; Young, DO; Ji, J; Shen, P. Involvement of JNK regulation in oxidative stress-mediated murine liver injury by microcystin-LR. Apoptosis 2008, 13, 1031–1042. [Google Scholar]
- Leiers, T; Bihlmayer, A; Ammon, HP; Wahl, MA. [Ca(2+)](i)- and insulin-stimulating effect of the non-membranepermeable phosphatase-inhibitor microcystin-LR in intact insulin-secreting cells (RINm5F). Br. J. Pharmacol 2000, 130, 1406–1410. [Google Scholar]
- Cnop, M; Welsh, N; Jonas, JC; Jorns, A; Lenzen, S; Eizirik, DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 2005, 54(Suppl 2), S97–107. [Google Scholar]
- Prentki, M; Segall, L; Roche, E; Thumelin, S; Brun, T; McGarry, JD; Corkey, BE; Assimacopoulos-Jeannet, F. Gluco-lipotoxicity and gene expression in the pancreatic beta cell. J Annu Diabetol Hotel Dieu 1998, 9773607, 17–27. [Google Scholar]
- McDaniel, ML; Corbett, JA; Kwon, G; Hill, JR. A role for nitric oxide and other inflammatory mediators in cytokine-induced pancreatic beta-cell dysfunction and destruction. Adv. Exp. Med. Biol 1997, 426, 313–319. [Google Scholar]
- Shimabukuro, M; Zhou, YT; Levi, M; Unger, RH. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar]
- Maedler, K; Sergeev, P; Ris, F; Oberholzer, J; Joller-Jemelka, HI; Spinas, GA; Kaiser, N; Halban, PA; Donath, MY. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest 2002, 110, 851–860. [Google Scholar]
- Fischer, A; Hoeger, SJ; Stemmer, K; Feurstein, DJ; Knobeloch, D; Nussler, A; Dietrich, DR. The role of organic anion transporting polypeptides (OATPs/SLCOs) in the toxicity of different microcystin congeners in vitro: A comparison of primary human hepatocytes and OATP-transfected HEK293 cells. Toxicol. Appl. Pharmacol 2010, 245, 9–20. [Google Scholar]
- Monks, NR; Liu, S; Xu, Y; Yu, H; Bendelow, AS; Moscow, JA. Potent cytotoxicity of the phosphatase inhibitor microcystin LR and microcystin analogues in OATP1B1- and OATP1B3-expressing HeLa cells. Mol. Cancer Ther 2007, 6, 587–598. [Google Scholar]
- Giacomini, KM; Huang, SM; Tweedie, DJ; Benet, LZ; Brouwer, KL; Chu, X; Dahlin, A; Evers, R; Fischer, V; Hillgren, KM; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov 2010, 9, 215–236. [Google Scholar]
- Chen, W; Song, L; Ou, D; Gan, N. Chronic toxicity and responses of several important enzymes in Daphnia magna on exposure to sublethal microcystin-LR. Environ. Toxicol 2005, 20, 323–330. [Google Scholar]



© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ji, Y.; Lu, G.; Chen, G.; Huang, B.; Zhang, X.; Shen, K.; Wu, S. Microcystin-LR Induces Apoptosis via NF-κB /iNOS Pathway in INS-1 Cells. Int. J. Mol. Sci. 2011, 12, 4722-4734. https://doi.org/10.3390/ijms12074722
Ji Y, Lu G, Chen G, Huang B, Zhang X, Shen K, Wu S. Microcystin-LR Induces Apoptosis via NF-κB /iNOS Pathway in INS-1 Cells. International Journal of Molecular Sciences. 2011; 12(7):4722-4734. https://doi.org/10.3390/ijms12074722
Chicago/Turabian StyleJi, Yong, Gao Lu, Guoqiang Chen, Bin Huang, Xian Zhang, Kai Shen, and Song Wu. 2011. "Microcystin-LR Induces Apoptosis via NF-κB /iNOS Pathway in INS-1 Cells" International Journal of Molecular Sciences 12, no. 7: 4722-4734. https://doi.org/10.3390/ijms12074722