What Do Effective Treatments for Multiple Sclerosis Tell Us about the Molecular Mechanisms Involved in Pathogenesis?

Abstract
:1. Introduction
1.1. T-Cells in MS
1.2. Regulatory T-Cells in MS
1.3. NK Cells in MS
1.4. B-Cells in MS
1.5. Monocytes and Macrophages in MS
1.6. Through the Looking Glass
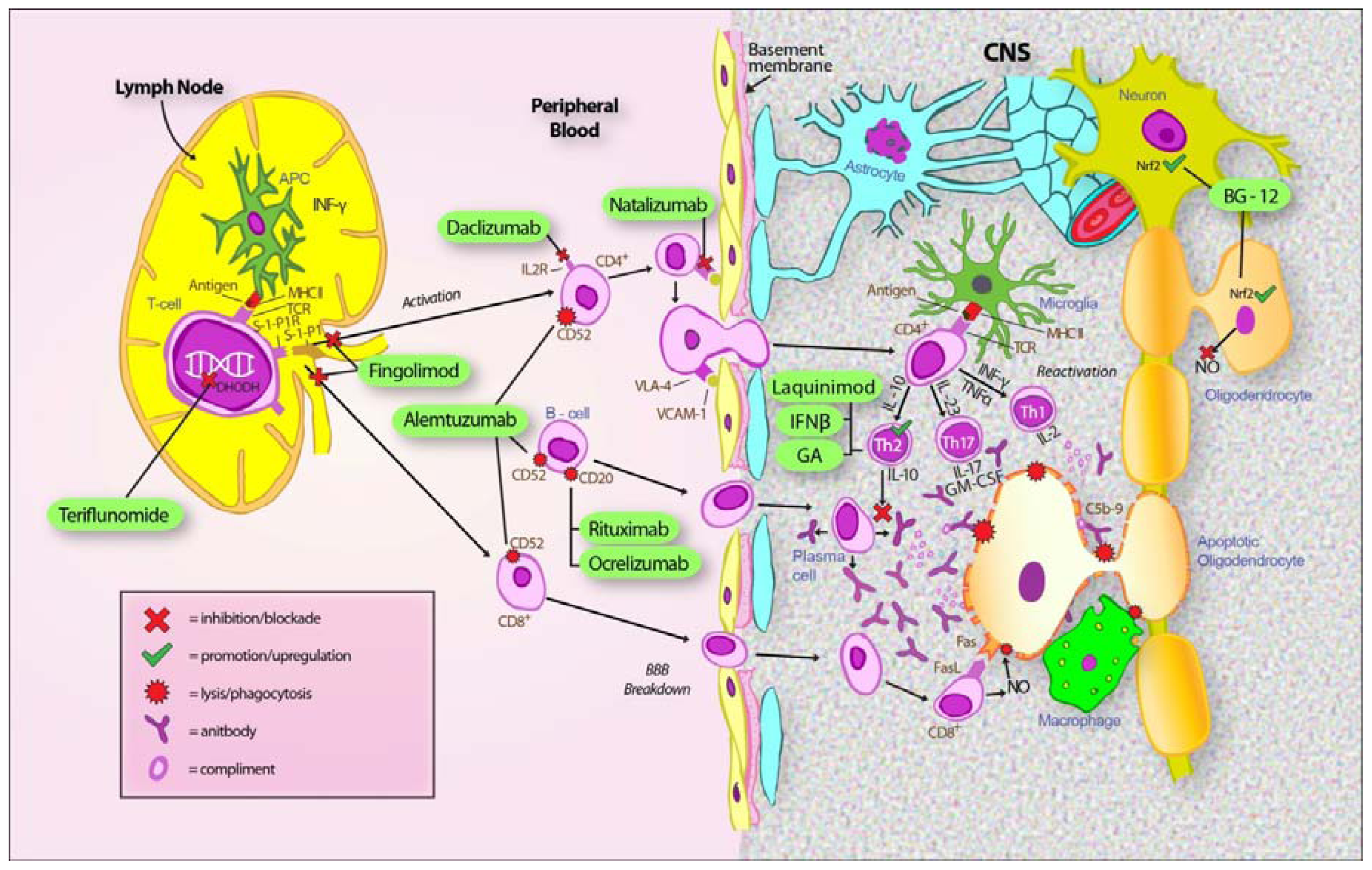
2. Approved Disease Modifying Treatment in RRMS
2.1. Interferons
2.2. Glatiramer Acetate
2.3. Natalizumab
2.4. Fingolimod
3. Emerging Disease Modifying Treatment in RRMS
3.1. Dimethyl Fumarate (BG-12)
3.2. Teriflunomide
3.3. Laquinimod
3.4. Alemtuzumab
3.5. Daclizumab
3.6. B-Cell Therapies
3.6.1. Rituximab
3.6.2. Ocrelizumab
3.6.3. Atacicept
4. Conclusions
Acknowledgments
References
- Clanet, M. Jean-Martin Charcot. 1825 to 1893. Int. MS J 2008, 15, 59–61. [Google Scholar]
- Orton, S.M.; Herrera, B.M.; Yee, I.M.; Valdar, W.; Ramagopalan, S.V.; Sadovnick, A.D.; Ebers, G.C. Sex ratio of multiple sclerosis in Canada: A longitudinal study. Lancet Neurol 2006, 5, 932–936. [Google Scholar]
- Olerup, O.; Hillert, J. HLA class II-associated genetic susceptibility in multiple sclerosis: A critical evaluation. Tissue Antigens 1991, 38, 1–15. [Google Scholar]
- Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; de Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; Ivinson, A.J.; et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med 2007, 357, 851–862. [Google Scholar]
- Weber, F.; Fontaine, B.; Cournu-Rebeix, I.; Kroner, A.; Knop, M.; Lutz, S.; Muller-Sarnowski, F.; Uhr, M.; Bettecken, T.; Kohli, M.; et al. IL2RA and IL7RA genes confer susceptibility for multiple sclerosis in two independent European populations. Genes Immun 2008, 9, 259–263. [Google Scholar]
- Stromnes, I.M.; Goverman, J.M. Active induction of experimental allergic encephalomyelitis. Nat. Protoc 2006, 1, 1810–1819. [Google Scholar]
- Wegner, C. Pathological differences in acute inflammatory demyelinating diseases of the central nervous system. Int. MS J 2005, 12, 13–19. [Google Scholar]
- Stromnes, I.M.; Goverman, J.M. Passive induction of experimental allergic encephalomyelitis. Nat. Protoc 2006, 1, 1952–1960. [Google Scholar]
- Steinman, L. Assessment of animal models for MS and demyelinating disease in the design of rational therapy. Neuron 1999, 24, 511–514. [Google Scholar]
- Molyneux, P.D.; Filippi, M.; Barkhof, F.; Gasperini, C.; Yousry, T.A.; Truyen, L.; Lai, H.M.; Rocca, M.A.; Moseley, I.F.; Miller, D.H. Correlations between monthly enhanced MRI lesion rate and changes in T2 lesion volume in multiple sclerosis. Ann. Neurol 1998, 43, 332–339. [Google Scholar]
- Grossman, R.I.; Gonzalez-Scarano, F.; Atlas, S.W.; Galetta, S.; Silberberg, D.H. Multiple sclerosis: Gadolinium enhancement in MR imaging. Radiology 1986, 161, 721–725. [Google Scholar]
- Moore, G.R.; Laule, C. Neuropathologic correlates of magnetic resonance imaging in multiple sclerosis. J. Neuropathol. Exp. Neurol 2012, 71, 762–778. [Google Scholar]
- McFarland, H.F.; Barkhof, F.; Antel, J.; Miller, D.H. The role of MRI as a surrogate outcome measure in multiple sclerosis. Mult. Scler 2002, 8, 40–51. [Google Scholar]
- Paty, D.W.; Li, D.K. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology 1993, 43, 662–667. [Google Scholar]
- Frank, J.A.; Stone, L.A.; Smith, M.E.; Albert, P.S.; Maloni, H.; McFarland, H.F. Serial contrast-enhanced magnetic resonance imaging in patients with early relapsing-remitting multiple sclerosis: Implications for treatment trials. Ann. Neurol 1994, 36, S86–S90. [Google Scholar]
- Broderick, J.P.; Narayan, S.; Gaskill, M.; Dhawan, A.P.; Khoury, J. Volumetric measurement of multifocal brain lesions. Implications for treatment trials of vascular dementia and multiple sclerosis. J. Neuroimaging 1996, 6, 36–43. [Google Scholar]
- Rudick, R.A.; Fisher, E.; Lee, J.C.; Duda, J.T.; Simon, J. Brain atrophy in relapsing multiple sclerosis: Relationship to relapses, EDSS, and treatment with interferon beta-1a. Mult. Scler 2000, 6, 365–372. [Google Scholar]
- Delves, P.J.; Roitt, I.M. The immune system. Second of two parts. N. Engl. J. Med 2000, 343, 108–117. [Google Scholar]
- Mosmann, T.R.; Sad, S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 1996, 17, 138–146. [Google Scholar]
- Steinman, L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat. Med 2007, 13, 139–145. [Google Scholar]
- Ferber, I.A.; Brocke, S.; Taylor-Edwards, C.; Ridgway, W.; Dinisco, C.; Steinman, L.; Dalton, D.; Fathman, C.G. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol 1996, 156, 5–7. [Google Scholar]
- Lin, J.; Ziring, D.; Desai, S.; Kim, S.; Wong, M.; Korin, Y.; Braun, J.; Reed, E.; Gjertson, D.; Singh, R.R. TNFalpha blockade in human diseases: An overview of efficacy and safety. Clin. Immunol 2008, 126, 13–30. [Google Scholar]
- The Lenercept Multiple Sclerosis Study Group; The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457–465.
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med 2005, 201, 233–240. [Google Scholar]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol 2011, 12, 568–575. [Google Scholar]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol 2011, 12, 560–567. [Google Scholar]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Pedras-Vasconcelos, J.; Verthelyi, D.; Dittel, B.N. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J. Immunol 2007, 178, 39–48. [Google Scholar]
- Hofstetter, H.H.; Ibrahim, S.M.; Koczan, D.; Kruse, N.; Weishaupt, A.; Toyka, K.V.; Gold, R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell. Immunol 2005, 237, 123–130. [Google Scholar]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol 2006, 177, 566–573. [Google Scholar]
- O’Connor, R.A.; Prendergast, C.T.; Sabatos, C.A.; Lau, C.W.; Leech, M.D.; Wraith, D.C.; Anderton, S.M. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J. Immunol 2008, 181, 3750–3754. [Google Scholar]
- Kang, Z.; Altuntas, C.Z.; Gulen, M.F.; Liu, C.; Giltiay, N.; Qin, H.; Liu, L.; Qian, W.; Ransohoff, R.M.; Bergmann, C.; et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 2010, 32, 414–425. [Google Scholar]
- Rodgers, J.M.; Zhou, L.; Miller, S.D. Act1, scene brain: Astrocytes play a lead role. Immunity 2010, 32, 302–304. [Google Scholar]
- Burfoot, R.K.; Jensen, C.J.; Field, J.; Stankovich, J.; Varney, M.D.; Johnson, L.J.; Butzkueven, H.; Booth, D.; Bahlo, M.; Tait, B.D.; et al. SNP mapping and candidate gene sequencing in the class I region of the HLA complex: Searching for multiple sclerosis susceptibility genes in Tasmanians. Tissue Antigens 2008, 71, 42–50. [Google Scholar]
- Friese, M.A.; Fugger, L. Pathogenic CD8(+) T cells in multiple sclerosis. Ann. Neurol 2009, 66, 132–141. [Google Scholar]
- Kuhlmann, T.; Lingfeld, G.; Bitsch, A.; Schuchardt, J.; Bruck, W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 2002, 125, 2202–2212. [Google Scholar]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol 2008, 172, 146–155. [Google Scholar]
- Anderson, A.C.; Chandwaskar, R.; Lee, D.H.; Sullivan, J.M.; Solomon, A.; Rodriguez-Manzanet, R.; Greve, B.; Sobel, R.A.; Kuchroo, V.K. A transgenic model of central nervous system autoimmunity mediated by CD4(+) and CD8(+) T and B cells. J. Immunol 2012, 188, 2084–2092. [Google Scholar]
- Kohm, A.P.; Carpentier, P.A.; Anger, H.A.; Miller, S.D. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol 2002, 169, 4712–4716. [Google Scholar]
- Huan, J.; Culbertson, N.; Spencer, L.; Bartholomew, R.; Burrows, G.G.; Chou, Y.K.; Bourdette, D.; Ziegler, S.F.; Offner, H.; Vandenbark, A.A. Decreased FOXP3 levels in multiple sclerosis patients. J. Neurosci. Res 2005, 81, 45–52. [Google Scholar]
- Dalla Libera, D.; di Mitri, D.; Bergami, A.; Centonze, D.; Gasperini, C.; Grasso, M.G.; Galgani, S.; Martinelli, V.; Comi, G.; Avolio, C.; et al. T regulatory cells are markers of disease activity in multiple sclerosis patients. PLoS One 2011, 6, e21386. [Google Scholar]
- Putheti, P.; Pettersson, A.; Soderstrom, M.; Link, H.; Huang, Y.M. Circulating CD4+CD25+ T regulatory cells are not altered in multiple sclerosis and unaffected by disease-modulating drugs. J. Clin. Immunol 2004, 24, 155–161. [Google Scholar]
- Michel, L.; Berthelot, L.; Pettre, S.; Wiertlewski, S.; Lefrere, F.; Braudeau, C.; Brouard, S.; Soulillou, J.P.; Laplaud, D.A. Patients with relapsing-remitting multiple sclerosis have normal Treg function when cells expressing IL-7 receptor alpha-chain are excluded from the analysis. J. Clin. Invest 2008, 118, 3411–3419. [Google Scholar]
- Viglietta, V.; Baecher-Allan, C.; Weiner, H.L.; Hafler, D.A. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med 2004, 199, 971–979. [Google Scholar]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001, 97, 3146–3151. [Google Scholar]
- Berzins, S.P.; Smyth, M.J.; Baxter, A.G. Presumed guilty: Natural killer T cell defects and human disease. Nat. Rev. Immunol 2011, 11, 131–142. [Google Scholar]
- Benczur, M.; Petranyl, G.G.; Palffy, G.; Varga, M.; Talas, M.; Kotsy, B.; Foldes, I.; Hollan, S.R. Dysfunction of natural killer cells in multiple sclerosis: A possible pathogenetic factor. Clin. Exp. Immunol 1980, 39, 657–662. [Google Scholar]
- Zhang, B.; Yamamura, T.; Kondo, T.; Fujiwara, M.; Tabira, T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J. Exp. Med 1997, 186, 1677–1687. [Google Scholar]
- Kaudewitz, P.; Zander, H.; Abb, J.; Ziegler-Heitbrock, H.W.; Riethmuller, G. Genetic influence on natural cytotoxicity and interferon production in multiple sclerosis studies in monozygotic discordant twins. Hum. Immunol 1983, 7, 51–58. [Google Scholar]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol 2000, 47, 707–717. [Google Scholar]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004, 14, 164–174. [Google Scholar]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130, 1089–1104. [Google Scholar]
- Lovato, L.; Willis, S.N.; Rodig, S.J.; Caron, T.; Almendinger, S.E.; Howell, O.W.; Reynolds, R.; O’Connor, K.C.; Hafler, D.A. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 2011, 134, 534–541. [Google Scholar]
- Lechner-Scott, J.; Spencer, B.; de Malmanche, T.; Attia, J.; Fitzgerald, M.; Trojano, M.; Grand’maison, F.; Gomez, J.A.; Izquierdo, G.; Duquette, P.; et al. The frequency of CSF oligoclonal banding in multiple sclerosis increases with latitude. Mult. Scler 2012, 18, 974–982. [Google Scholar]
- Obermeier, B.; Lovato, L.; Mentele, R.; Bruck, W.; Forne, I.; Imhof, A.; Lottspeich, F.; Turk, K.W.; Willis, S.N.; Wekerle, H.; et al. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J. Neuroimmunol 2011, 233, 245–248. [Google Scholar]
- Bernard, C.C.; de Rosbo, N.K. Immunopathological recognition of autoantigens in multiple sclerosis. Acta Neurol. (Napoli) 1991, 13, 171–178. [Google Scholar]
- Owens, G.P.; Bennett, J.L.; Lassmann, H.; O’Connor, K.C.; Ritchie, A.M.; Shearer, A.; Lam, C.; Yu, X.; Birlea, M.; DuPree, C.; et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann. Neurol 2009, 65, 639–649. [Google Scholar]
- Genain, C.P.; Cannella, B.; Hauser, S.L.; Raine, C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med 1999, 5, 170–175. [Google Scholar]
- Wootla, B.; Denic, A.; Keegan, B.M.; Winters, J.L.; Astapenko, D.; Warrington, A.E.; Bieber, A.J.; Rodriguez, M. Evidence for the role of B cells and immunoglobulins in the pathogenesis of multiple sclerosis. Neurol. Res. Int 2011, 2011, 780712. [Google Scholar]
- Disanto, G.; Morahan, J.M.; Barnett, M.H.; Giovannoni, G.; Ramagopalan, S.V. The evidence for a role of B cells in multiple sclerosis. Neurology 2012, 78, 823–832. [Google Scholar]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med 2004, 350, 1328–1337. [Google Scholar]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol 2007, 61, 288–299. [Google Scholar]
- Thacker, E.L.; Mirzaei, F.; Ascherio, A. Infectious mononucleosis and risk for multiple sclerosis: A meta-analysis. Ann. Neurol 2006, 59, 499–503. [Google Scholar]
- Farrell, R.A.; Antony, D.; Wall, G.R.; Clark, D.A.; Fisniku, L.; Swanton, J.; Khaleeli, Z.; Schmierer, K.; Miller, D.H.; Giovannoni, G. Humoral immune response to EBV in multiple sclerosis is associated with disease activity on MRI. Neurology 2009, 73, 32–38. [Google Scholar]
- Pender, M.P. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol 2003, 24, 584–588. [Google Scholar]
- Pender, M.P. CD8+ T-Cell deficiency, epstein-barr virus infection, vitamin D deficiency, and steps to autoimmunity: A unifying hypothesis. Autoimmune Dis 2012, 2012, 189096. [Google Scholar]
- Tzartos, J.S.; Khan, G.; Vossenkamper, A.; Cruz-Sadaba, M.; Lonardi, S.; Sefia, E.; Meager, A.; Elia, A.; Middeldorp, J.M.; Clemens, M.; et al. Association of innate immune activation with latent Epstein-Barr virus in active MS lesions. Neurology 2012, 78, 15–23. [Google Scholar]
- Serafini, B.; Rosicarelli, B.; Franciotta, D.; Magliozzi, R.; Reynolds, R.; Cinque, P.; Andreoni, L.; Trivedi, P.; Salvetti, M.; Faggioni, A.; et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J. Exp. Med 2007, 204, 2899–2912. [Google Scholar]
- Willis, S.N.; Stadelmann, C.; Rodig, S.J.; Caron, T.; Gattenloehner, S.; Mallozzi, S.S.; Roughan, J.E.; Almendinger, S.E.; Blewett, M.M.; Bruck, W.; et al. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain 2009, 132, 3318–3328. [Google Scholar]
- Sargsyan, S.A.; Shearer, A.J.; Ritchie, A.M.; Burgoon, M.P.; Anderson, S.; Hemmer, B.; Stadelmann, C.; Gattenlohner, S.; Owens, G.P.; Gilden, D.; et al. Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology 2010, 74, 1127–1135. [Google Scholar]
- Lang, H.L.; Jacobsen, H.; Ikemizu, S.; Andersson, C.; Harlos, K.; Madsen, L.; Hjorth, P.; Sondergaard, L.; Svejgaard, A.; Wucherpfennig, K.; et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol 2002, 3, 940–943. [Google Scholar]
- Miller, G. Immortalization of human lymphocytes by Epstein-Barr virus. Yale J. Biol. Med 1982, 55, 305–310. [Google Scholar]
- Ray, A.; Mann, M.K.; Basu, S.; Dittel, B.N. A case for regulatory B cells in controlling the severity of autoimmune-mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J. Neuroimmunol 2011, 230, 1–9. [Google Scholar]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med 2008, 358, 676–688. [Google Scholar]
- Bar-Or, A.; Calabresi, P.A.; Arnold, D.; Markowitz, C.; Shafer, S.; Kasper, L.H.; Waubant, E.; Gazda, S.; Fox, R.J.; Panzara, M.; et al. Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Ann. Neurol 2008, 63, 395–400. [Google Scholar]
- Naismith, R.T.; Piccio, L.; Lyons, J.A.; Lauber, J.; Tutlam, N.T.; Parks, B.J.; Trinkaus, K.; Song, S.K.; Cross, A.H. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: A 52-week phase II trial. Neurology 2010, 74, 1860–1867. [Google Scholar]
- Harp, C.T.; Lovett-Racke, A.E.; Racke, M.K.; Frohman, E.M.; Monson, N.L. Impact of myelin-specific antigen presenting B cells on T cell activation in multiple sclerosis. Clin. Immunol 2008, 128, 382–391. [Google Scholar]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann. Neurol 2010, 67, 452–461. [Google Scholar]
- Benveniste, E.N. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J. Mol. Med. (Berl) 1997, 75, 165–173. [Google Scholar]
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal damage in acute multiple sclerosis lesions. Brain 1997, 120, 393–399. [Google Scholar]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar]
- Xiao, B.G.; Link, H. Is there a balance between microglia and astrocytes in regulating Th1/Th2-cell responses and neuropathologies? Immunol. Today 1999, 20, 477–479. [Google Scholar]
- De Simone, R.; Giampaolo, A.; Giometto, B.; Gallo, P.; Levi, G.; Peschle, C.; Aloisi, F. The costimulatory molecule B7 is expressed on human microglia in culture and in multiple sclerosis acute lesions. J. Neuropathol. Exp. Neurol 1995, 54, 175–187. [Google Scholar]
- Cannella, B.; Raine, C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol 1995, 37, 424–435. [Google Scholar]
- Chao, C.C.; Hu, S.; Sheng, W.S.; Peterson, P.K. Tumor necrosis factor-alpha production by human fetal microglial cells: Regulation by other cytokines. Dev. Neurosci 1995, 17, 97–105. [Google Scholar]
- Merrill, J.E.; Ignarro, L.J.; Sherman, M.P.; Melinek, J.; Lane, T.E. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J. Immunol 1993, 151, 2132–2141. [Google Scholar]
- Prineas, J.W.; Wright, R.G. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab. Invest 1978, 38, 409–421. [Google Scholar]
- Dalakas, M.; Wright, R.G.; Prineas, J.W. Nature of the reversible white matter lesion in multiple sclerosis. Effects of acute inflammation on myelinated tissue studied in the rabbit eye. Brain 1980, 103, 515–524. [Google Scholar]
- Walsh, M.J.; Murray, J.M. Dual implication of 2′,3′-cyclic nucleotide 3′ phosphodiesterase as major autoantigen and C3 complement-binding protein in the pathogenesis of multiple sclerosis. J. Clin. Invest 1998, 101, 1923–1931. [Google Scholar]
- Langer-Gould, A.; Atlas, S.W.; Green, A.J.; Bollen, A.W.; Pelletier, D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med 2005, 353, 375–381. [Google Scholar]
- Jacobs, L.D.; Cookfair, D.L.; Rudick, R.A.; Herndon, R.M.; Richert, J.R.; Salazar, A.M.; Fischer, J.S.; Goodkin, D.E.; Granger, C.V.; Simon, J.H.; et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann. Neurol 1996, 39, 285–294. [Google Scholar]
- Ebers, G.C. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet 1998, 352, 1498–1504. [Google Scholar]
- The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology 1993, 43, 655–661.
- Rudick, R.A.; Goodkin, D.E.; Jacobs, L.D.; Cookfair, D.L.; Herndon, R.M.; Richert, J.R.; Salazar, A.M.; Fischer, J.S.; Granger, C.V.; Simon, J.H.; et al. Impact of interferon beta-1a on neurologic disability in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Neurology 1997, 49, 358–363. [Google Scholar]
- Johnson, K.P.; Brooks, B.R.; Cohen, J.A.; Ford, C.C.; Goldstein, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Rose, J.W.; Schiffer, R.B.; et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: Results of a phase III multicenter, double-blind placebo-controlled trial. Neurology 1995, 45, 1268–1276. [Google Scholar]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med 2006, 354, 899–910. [Google Scholar]
- Kappos, L.; Radue, E.W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med 2010, 362, 387–401. [Google Scholar]
- Khatri, B.; Barkhof, F.; Comi, G.; Hartung, H.P.; Kappos, L.; Montalban, X.; Pelletier, J.; Stites, T.; Wu, S.; Holdbrook, F.; et al. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: A randomised extension of the TRANSFORMS study. Lancet Neurol 2011, 10, 520–529. [Google Scholar]
- Fox, R. Clinical Efficacy of BG-12 in Relapsing-Remitting Multiple Sclerosis (RRMS): Data from the Phase 3 CONFIRM Study. Available online: http://www.abstracts2view.com/aan/view.php?nu=AAN12L_S01_003 accessed on 18 September 2012.
- Gold, R. Clinical Efficacy of BG-12, An Oral Therapy, in Relapsing-Remitting Multiple Sclerosis: Data from the Phase 3 DEFINE Trial. Available online: http://registration.akm.ch/einsicht.php?XNABSTRACT_ID=137258&XNSPRACHE_ID=2&XNKONGRESS_ID=150&XNMASKEN_ID=900 accessed on 18 September 2012.
- Phillips, J.T.; Fox, L.M.; Miller, D.C.; Kita, M.; Hutchinson, M.; Havrdova, E.; Raghupathi, K.; Yuan, H.; Novas, M.; Viglietta, V.; et al. Safety and Tolerability of BG-12 in Patients with Relapsing-Remitting Multiple Sclerosis (RRMS): Analyses from the CONFIRM Study. Proceedings of American Academy of Neurology, New Orleans, LA, USA, 21–28 April 2012.
- O’Connor, P.; Wolinsky, J.S.; Confavreux, C.; Comi, G.; Kappos, L.; Olsson, T.P.; Benzerdjeb, H.; Truffinet, P.; Wang, L.; Miller, A.; et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N. Engl. J. Med 2011, 365, 1293–1303. [Google Scholar]
- Comi, G.; Jeffery, D.; Kappos, L.; Montalban, X.; Boyko, A.; Rocca, M.A.; Filippi, M. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N. Engl. J. Med 2012, 366, 1000–1009. [Google Scholar]
- Cohen, J.A. Efficacy and Safety Results from Comparison of Alemtuzumab and Rebif® Efficacy in Multiple Sclerosis II (CARE-MS II): A Phase 3 Study in Relapsing-Remitting Multiple Sclerosis Patients Who Relapsed on Prior Therapy. Proceedings of American Academy of Neurology (AAN), New Orleans, LA, USA, 21–28 April,2012; S01.004.
- Coles, A.J. Efficacy and Safety Results from Comparison of Alemtuzumab and Rebif® Efficacy in Multiple Sclerosis I (CARE-MS I): A Phase 3 Study in Relapsing-Remitting Treatment-Naïve Patients. Proceedings of American Academy of Neurology, New Orleans, LA, USA, 21–28 April,2012; S01.006.
- Goodin, D.S.; Jones, J.; Li, D.; Traboulsee, A.; Reder, A.T.; Beckmann, K.; Konieczny, A.; Knappertz, V. Establishing long-term efficacy in chronic disease: Use of recursive partitioning and propensity score adjustment to estimate outcome in MS. PLoS One 2011, 6, e22444. [Google Scholar]
- Shirani, A.; Zhao, Y.; Karim, M.E.; Evans, C.; Kingwell, E.; van der Kop, M.L.; Oger, J.; Gustafson, P.; Petkau, J.; Tremlett, H. Association between use of interferon beta and progression of disability in patients with relapsing-remitting multiple sclerosis. JAMA 2012, 308, 247–256. [Google Scholar]
- McCoy, L.; Tsunoda, I.; Fujinami, R.S. Multiple sclerosis and virus induced immune responses: Autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 2006, 39, 9–19. [Google Scholar]
- Chadha, K.C.; Ambrus, J.L., Jr; Dembinski, W.; Ambrus, J.L., Sr. Interferons and interferon inhibitory activity in disease and therapy. Exp. Biol. Med. (Maywood) 2004, 229, 285–290. [Google Scholar]
- Kozovska, M.E.; Hong, J.; Zang, Y.C.; Li, S.; Rivera, V.M.; Killian, J.M.; Zhang, J.Z. Interferon beta induces T-helper 2 immune deviation in MS. Neurology 1999, 53, 1692–1697. [Google Scholar]
- Liu, Z.; Pelfrey, C.M.; Cotleur, A.; Lee, J.C.; Rudick, R.A. Immunomodulatory effects of interferon beta-1a in multiple sclerosis. J. Neuroimmunol 2001, 112, 153–162. [Google Scholar]
- Ozenci, V.; Kouwenhoven, M.; Huang, Y.M.; Xiao, B.; Kivisakk, P.; Fredrikson, S.; Link, H. Multiple sclerosis: Levels of interleukin-10-secreting blood mononuclear cells are low in untreated patients but augmented during interferon-beta-1b treatment. Scand. J. Immunol 1999, 49, 554–561. [Google Scholar]
- Chen, M.; Chen, G.; Nie, H.; Zhang, X.; Niu, X.; Zang, Y.C.; Skinner, S.M.; Zhang, J.Z.; Killian, J.M.; Hong, J. Regulatory effects of IFN-beta on production of osteopontin and IL-17 by CD4+ T Cells in MS. Eur. J. Immunol 2009, 39, 2525–2536. [Google Scholar]
- Zhang, L.; Yuan, S.; Cheng, G.; Guo, B. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One 2011, 6, e28432. [Google Scholar]
- Guo, B.; Chang, E.Y.; Cheng, G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J. Clin. Invest 2008, 118, 1680–1690. [Google Scholar]
- Shinohara, M.L.; Kim, J.H.; Garcia, V.A.; Cantor, H. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: Role of intracellular osteopontin. Immunity 2008, 29, 68–78. [Google Scholar]
- Ramgolam, V.S.; Sha, Y.; Jin, J.; Zhang, X.; Markovic-Plese, S. IFN-beta inhibits human Th17 cell differentiation. J. Immunol 2009, 183, 5418–5427. [Google Scholar]
- Kieseier, B.C. The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs 2011, 25, 491–502. [Google Scholar]
- Saraste, M.; Irjala, H.; Airas, L. Expansion of CD56Bright natural killer cells in the peripheral blood of multiple sclerosis patients treated with interferon-beta. Neurol. Sci 2007, 28, 121–126. [Google Scholar]
- Vandenbark, A.A.; Huan, J.; Agotsch, M.; La Tocha, D.; Goelz, S.; Offner, H.; Lanker, S.; Bourdette, D. Interferon-beta-1a treatment increases CD56bright natural killer cells and CD4+CD25+ Foxp3 expression in subjects with multiple sclerosis. J. Neuroimmunol 2009, 215, 125–128. [Google Scholar]
- Sanvito, L.; Tomita, A.; Chihara, N.; Okamoto, T.; Lin, Y.; Ogawa, M.; Gran, B.; Aranami, T.; Yamamura, T. Increase of Ki-67+ natural killer cells in multiple sclerosis patients treated with interferon-beta and interferon-beta combined with low-dose oral steroids. J. Neuroimmunol 2011, 236, 111–117. [Google Scholar]
- Martinez-Rodriguez, J.E.; Lopez-Botet, M.; Munteis, E.; Rio, J.; Roquer, J.; Montalban, X.; Comabella, M. Natural killer cell phenotype and clinical response to interferon-beta therapy in multiple sclerosis. Clin. Immunol 2011, 141, 348–356. [Google Scholar]
- De Andres, C.; Aristimuno, C.; de Las Heras, V.; Martinez-Gines, M.L.; Bartolome, M.; Arroyo, R.; Navarro, J.; Gimenez-Roldan, S.; Fernandez-Cruz, E.; Sanchez-Ramon, S. Interferon beta-1a therapy enhances CD4+ regulatory T-cell function: An ex vivo and in vitro longitudinal study in relapsing-remitting multiple sclerosis. J. Neuroimmunol 2007, 182, 204–211. [Google Scholar]
- Korporal, M.; Haas, J.; Balint, B.; Fritzsching, B.; Schwarz, A.; Moeller, S.; Fritz, B.; Suri-Payer, E.; Wildemann, B. Interferon beta-induced restoration of regulatory T-cell function in multiple sclerosis is prompted by an increase in newly generated naive regulatory T cells. Arch. Neurol 2008, 65, 1434–1439. [Google Scholar]
- Chen, M.; Chen, G.; Deng, S.; Liu, X.; Hutton, G.J.; Hong, J. IFN-beta induces the proliferation of CD4+CD25+Foxp3+ regulatory T cells through upregulation of GITRL on dendritic cells in the treatment of multiple sclerosis. J. Neuroimmunol 2012, 242, 39–46. [Google Scholar]
- Caggiula, M.; Batocchi, A.P.; Frisullo, G.; Angelucci, F.; Patanella, A.K.; Sancricca, C.; Nociti, V.; Tonali, P.A.; Mirabella, M. Neurotrophic factors in relapsing remitting and secondary progressive multiple sclerosis patients during interferon beta therapy. Clin. Immunol 2006, 118, 77–82. [Google Scholar]
- Boutros, T.; Croze, E.; Yong, V.W. Interferon-beta is a potent promoter of nerve growth factor production by astrocytes. J. Neurochem 1997, 69, 939–946. [Google Scholar]
- Biernacki, K.; Antel, J.P.; Blain, M.; Narayanan, S.; Arnold, D.L.; Prat, A. Interferon beta promotes nerve growth factor secretion early in the course of multiple sclerosis. Arch. Neurol 2005, 62, 563–568. [Google Scholar]
- Ramgolam, V.S.; Sha, Y.; Marcus, K.L.; Choudhary, N.; Troiani, L.; Chopra, M.; Markovic-Plese, S. B cells as a therapeutic target for IFN-beta in relapsing-remitting multiple sclerosis. J. Immunol 2011, 186, 4518–4526. [Google Scholar]
- Ebers, G.C.; Reder, A.T.; Traboulsee, A.; Li, D.; Langdon, D.; Goodin, D.S.; Wolf, C.; Beckmann, K.; Konieczny, A. Long-term follow-up of the original interferon-beta1b trial in multiple sclerosis: Design and lessons from a 16-year observational study. Clin. Ther 2009, 31, 1724–1736. [Google Scholar]
- Kim, S.H.; Kim, W.; Li, X.F.; Jung, I.J.; Kim, H.J. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult. Scler 2012. [Google Scholar] [CrossRef]
- Axtell, R.C.; de Jong, B.A.; Boniface, K.; van der Voort, L.F.; Bhat, R.; de Sarno, P.; Naves, R.; Han, M.; Zhong, F.; Castellanos, J.G.; et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat. Med 2010, 16, 406–412. [Google Scholar]
- Bushnell, S.E.; Zhao, Z.; Stebbins, C.C.; Cadavid, D.; Buko, A.M.; Whalley, E.T.; Davis, J.A.; Versage, E.M.; Richert, J.R.; Axtell, R.C.; et al. Serum IL-17F does not predict poor response to IM IFNbeta-1a in relapsing-remitting MS. Neurology 2012, 79, 531–537. [Google Scholar]
- Sela, M.; Teitelbaum, D. Glatiramer acetate in the treatment of multiple sclerosis. Expert Opin. Pharmacother 2001, 2, 1149–1165. [Google Scholar]
- Bornstein, M.B.; Miller, A.; Slagle, S.; Weitzman, M.; Crystal, H.; Drexler, E.; Keilson, M.; Merriam, A.; Wassertheil-Smoller, S.; Spada, V.; et al. A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis. N. Engl. J. Med 1987, 317, 408–414. [Google Scholar]
- Comi, G.; Filippi, M.; Wolinsky, J.S. European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects of glatiramer acetate on magnetic resonance imaging-measured disease activity and burden in patients with relapsing multiple sclerosis. Ann. Neurol 2001, 49, 290–297. [Google Scholar]
- Mikol, D.D.; Barkhof, F.; Chang, P.; Coyle, P.K.; Jeffery, D.R.; Schwid, S.R.; Stubinski, B.; Uitdehaag, B.M. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): A multicentre, randomised, parallel, open-label trial. Lancet Neurol 2008, 7, 903–914. [Google Scholar]
- O’Connor, P.; Filippi, M.; Arnason, B.; Comi, G.; Cook, S.; Goodin, D.; Hartung, H.P.; Jeffery, D.; Kappos, L.; Boateng, F.; et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: A prospective, randomised, multicentre study. Lancet Neurol 2009, 8, 889–897. [Google Scholar]
- Kala, M.; Miravalle, A.; Vollmer, T. Recent insights into the mechanism of action of glatiramer acetate. J. Neuroimmunol 2011, 235, 9–17. [Google Scholar]
- Vieira, P.L.; Heystek, H.C.; Wormmeester, J.; Wierenga, E.A.; Kapsenberg, M.L. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J. Immunol 2003, 170, 4483–4488. [Google Scholar]
- Aharoni, R.; Eilam, R.; Stock, A.; Vainshtein, A.; Shezen, E.; Gal, H.; Friedman, N.; Arnon, R. Glatiramer acetate reduces Th-17 inflammation and induces regulatory T-cells in the CNS of mice with relapsing-remitting or chronic EAE. J. Neuroimmunol 2010, 225, 100–111. [Google Scholar]
- Jee, Y.; Liu, R.; Bai, X.F.; Campagnolo, D.I.; Shi, F.D.; Vollmer, T.L. Do Th2 cells mediate the effects of glatiramer acetate in experimental autoimmune encephalomyelitis? Int. Immunol 2006, 18, 537–544. [Google Scholar]
- Tennakoon, D.K.; Mehta, R.S.; Ortega, S.B.; Bhoj, V.; Racke, M.K.; Karandikar, N.J. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J. Immunol 2006, 176, 7119–7129. [Google Scholar]
- Karandikar, N.J.; Crawford, M.P.; Yan, X.; Ratts, R.B.; Brenchley, J.M.; Ambrozak, D.R.; Lovett-Racke, A.E.; Frohman, E.M.; Stastny, P.; Douek, D.C.; et al. Glatiramer acetate (Copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J. Clin. Invest 2002, 109, 641–649. [Google Scholar]
- Kala, M.; Rhodes, S.N.; Piao, W.H.; Shi, F.D.; Campagnolo, D.I.; Vollmer, T.L. B cells from glatiramer acetate-treated mice suppress experimental autoimmune encephalomyelitis. Exp. Neurol 2010, 221, 136–145. [Google Scholar]
- Begum-Haque, S.; Sharma, A.; Christy, M.; Lentini, T.; Ochoa-Reparaz, J.; Fayed, I.F.; Mielcarz, D.; Haque, A.; Kasper, L.H. Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol 2010, 219, 47–53. [Google Scholar]
- Begum-Haque, S.; Christy, M.; Ochoa-Reparaz, J.; Nowak, E.C.; Mielcarz, D.; Haque, A.; Kasper, L.H. Augmentation of regulatory B cell activity in experimental allergic encephalomyelitis by glatiramer acetate. J. Neuroimmunol 2011, 232, 136–144. [Google Scholar]
- Lalive, P.H.; Neuhaus, O.; Benkhoucha, M.; Burger, D.; Hohlfeld, R.; Zamvil, S.S.; Weber, M.S. Glatiramer acetate in the treatment of multiple sclerosis: Emerging concepts regarding its mechanism of action. CNS Drugs 2011, 25, 401–414. [Google Scholar]
- Weber, M.S.; Prod’homme, T.; Youssef, S.; Dunn, S.E.; Rundle, C.D.; Lee, L.; Patarroyo, J.C.; Stuve, O.; Sobel, R.A.; Steinman, L.; et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat. Med 2007, 13, 935–943. [Google Scholar]
- Toker, A.; Slaney, C.Y.; Backstrom, B.T.; Harper, J.L. Glatiramer acetate treatment directly targets CD11b(+)Ly6G(−) monocytes and enhances the suppression of autoreactive T cells in experimental autoimmune encephalomyelitis. Scand. J. Immunol 2011, 74, 235–243. [Google Scholar]
- Sarchielli, P.; Zaffaroni, M.; Floridi, A.; Greco, L.; Candeliere, A.; Mattioni, A.; Tenaglia, S.; di Filippo, M.; Calabresi, P. Production of brain-derived neurotrophic factor by mononuclear cells of patients with multiple sclerosis treated with glatiramer acetate, interferon-beta 1a, and high doses of immunoglobulins. Mult. Scler 2007, 13, 313–331. [Google Scholar]
- Blanco, Y.; Moral, E.A.; Costa, M.; Gomez-Choco, M.; Torres-Peraza, J.F.; Alonso-Magdalena, L.; Alberch, J.; Jaraquemada, D.; Arbizu, T.; Graus, F.; et al. Effect of glatiramer acetate (Copaxone) on the immunophenotypic and cytokine profile and BDNF production in multiple sclerosis: A longitudinal study. Neurosci. Lett 2006, 406, 270–275. [Google Scholar]
- Aharoni, R.; Eilam, R.; Domev, H.; Labunskay, G.; Sela, M.; Arnon, R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc. Natl. Acad. Sci. USA 2005, 102, 19045–19050. [Google Scholar]
- Azoulay, D.; Vachapova, V.; Shihman, B.; Miler, A.; Karni, A. Lower brain-derived neurotrophic factor in serum of relapsing remitting MS: Reversal by glatiramer acetate. J. Neuroimmunol 2005, 167, 215–218. [Google Scholar]
- Ziemssen, T.; Kumpfel, T.; Schneider, H.; Klinkert, W.E.; Neuhaus, O.; Hohlfeld, R. Secretion of brain-derived neurotrophic factor by glatiramer acetate-reactive T-helper cell lines: Implications for multiple sclerosis therapy. J. Neurol. Sci 2005, 233, 109–112. [Google Scholar]
- Pucci, E.; Giuliani, G.; Solari, A.; Simi, S.; Minozzi, S.; di Pietrantonj, C.; Galea, I. Natalizumab for relapsing remitting multiple sclerosis. Cochrane Database Syst. Rev 2011, CD007621. [Google Scholar]
- Bloomgren, G.; Richman, S.; Hotermans, C.; Subramanyam, M.; Goelz, S.; Natarajan, A.; Lee, S.; Plavina, T.; Scanlon, J.V.; Sandrock, A.; et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N. Engl. J. Med 2012, 366, 1870–1880. [Google Scholar]
- Biogen Idec Tysabri Safety Update. Available online: http://www.tapp.com.au/members/Tysabri_Safety_Update_160812.pdf accessed on 23 August 2012.
- Weiner, L.P.; Herndon, R.M.; Narayan, O.; Johnson, R.T.; Shah, K.; Rubinstein, L.J.; Preziosi, T.J.; Conley, F.K. Isolation of virus related to SV40 from patients with progressive multifocal leukoencephalopathy. N. Engl. J. Med 1972, 286, 385–390. [Google Scholar]
- Major, E.O.; Ault, G.S. Progressive multifocal leukoencephalopathy: Clinical and laboratory observations on a viral induced demyelinating disease in the immunodeficient patient. Curr. Opin. Neurol 1995, 8, 184–190. [Google Scholar]
- Kappos, L.; Bates, D.; Edan, G.; Eraksoy, M.; Garcia-Merino, A.; Grigoriadis, N.; Hartung, H.P.; Havrdova, E.; Hillert, J.; Hohlfeld, R.; et al. Natalizumab treatment for multiple sclerosis: Updated recommendations for patient selection and monitoring. Lancet Neurol 2011, 10, 745–758. [Google Scholar]
- Vellinga, M.M.; Castelijns, J.A.; Barkhof, F.; Uitdehaag, B.M.; Polman, C.H. Postwithdrawal rebound increase in T2 lesional activity in natalizumab-treated MS patients. Neurology 2008, 70, 1150–1151. [Google Scholar]
- Stuve, O.; Cravens, P.D.; Frohman, E.M.; Phillips, J.T.; Remington, G.M.; von Geldern, G.; Cepok, S.; Singh, M.P.; Tervaert, J.W.; de Baets, M.; et al. Immunologic, clinical, and radiologic status 14 months after cessation of natalizumab therapy. Neurology 2009, 72, 396–401. [Google Scholar]
- Rice, G.P.; Hartung, H.P.; Calabresi, P.A. Anti-alpha4 integrin therapy for multiple sclerosis: Mechanisms and rationale. Neurology 2005, 64, 1336–1342. [Google Scholar]
- Sato, T.; Tachibana, K.; Nojima, Y.; D’Avirro, N.; Morimoto, C. Role of the VLA-4 molecule in T cell costimulation. Identification of the tyrosine phosphorylation pattern induced by the ligation of VLA-4. J. Immunol 1995, 155, 2938–2947. [Google Scholar]
- Ramos-Cejudo, J.; Oreja-Guevara, C.; Stark Aroeira, L.; Rodriguez de Antonio, L.; Chamorro, B.; Diez-Tejedor, E. Treatment with natalizumab in relapsing-remitting multiple sclerosis patients induces changes in inflammatory mechanism. J. Clin. Immunol 2011, 31, 623–631. [Google Scholar]
- Skarica, M.; Eckstein, C.; Whartenby, K.A.; Calabresi, P.A. Novel mechanisms of immune modulation of natalizumab in multiple sclerosis patients. J. Neuroimmunol 2011, 235, 70–76. [Google Scholar]
- Kivisakk, P.; Healy, B.C.; Viglietta, V.; Quintana, F.J.; Hootstein, M.A.; Weiner, H.L.; Khoury, S.J. Natalizumab treatment is associated with peripheral sequestration of proinflammatory T cells. Neurology 2009, 72, 1922–1930. [Google Scholar]
- Bar-Or, A.; Nuttall, R.K.; Duddy, M.; Alter, A.; Kim, H.J.; Ifergan, I.; Pennington, C.J.; Bourgoin, P.; Edwards, D.R.; Yong, V.W. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain 2003, 126, 2738–2749. [Google Scholar]
- Chun, J.; Brinkmann, V. A mechanistically novel, first oral therapy for multiple sclerosis: The development of fingolimod (FTY720, Gilenya). Discov. Med 2011, 12, 213–228. [Google Scholar]
- Fujino, M.; Funeshima, N.; Kitazawa, Y.; Kimura, H.; Amemiya, H.; Suzuki, S.; Li, X.K. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J. Pharmacol. Exp. Ther 2003, 305, 70–77. [Google Scholar]
- Kataoka, H.; Sugahara, K.; Shimano, K.; Teshima, K.; Koyama, M.; Fukunari, A.; Chiba, K. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell. Mol. Immunol 2005, 2, 439–448. [Google Scholar]
- Webb, M.; Tham, C.S.; Lin, F.F.; Lariosa-Willingham, K.; Yu, N.; Hale, J.; Mandala, S.; Chun, J.; Rao, T.S. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J. Neuroimmunol 2004, 153, 108–121. [Google Scholar]
- Cohen, J.A.; Chun, J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann. Neurol 2011, 69, 759–777. [Google Scholar]
- Mehling, M.; Lindberg, R.; Raulf, F.; Kuhle, J.; Hess, C.; Kappos, L.; Brinkmann, V. Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology 2010, 75, 403–410. [Google Scholar]
- Kivisakk, P.; Mahad, D.J.; Callahan, M.K.; Sikora, K.; Trebst, C.; Tucky, B.; Wujek, J.; Ravid, R.; Staugaitis, S.M.; Lassmann, H.; et al. Expression of CCR7 in multiple sclerosis: Implications for CNS immunity. Ann. Neurol 2004, 55, 627–638. [Google Scholar]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar]
- Altmeyer, P.J.; Matthes, U.; Pawlak, F.; Hoffmann, K.; Frosch, P.J.; Ruppert, P.; Wassilew, S.W.; Horn, T.; Kreysel, H.W.; Lutz, G.; et al. Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J. Am. Acad. Dermarol 1994, 30, 977–981. [Google Scholar]
- Moharregh-Khiabani, D.; Linker, R.A.; Gold, R.; Stangel, M. Fumaric Acid and its esters: An emerging treatment for multiple sclerosis. Curr. Neuropharmacol 2009, 7, 60–64. [Google Scholar]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev 2012, 26, 203–234. [Google Scholar]
- Schon, M.P.; Boehncke, W.H. Psoriasis. N. Engl. J. Med 2005, 352, 1899–1912. [Google Scholar]
- de Jong, R.; Bezemer, A.C.; Zomerdijk, T.P.; van de Pouw-Kraan, T.; Ottenhoff, T.H.; Nibbering, P.H. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur. J. Immunol 1996, 26, 2067–2074. [Google Scholar]
- Treumer, F.; Zhu, K.; Glaser, R.; Mrowietz, U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J. Invest. Dermatol 2003, 121, 1383–1388. [Google Scholar]
- Schilling, S.; Goelz, S.; Linker, R.; Luehder, F.; Gold, R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol 2006, 145, 101–107. [Google Scholar]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar]
- Scannevin, R.H.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther 2012, 341, 274–284. [Google Scholar]
- Lin, S.X.; Lisi, L.; Dello Russo, C.; Polak, P.E.; Sharp, A.; Weinberg, G.; Kalinin, S.; Feinstein, D.L. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro 2011, 3. [Google Scholar] [CrossRef]
- Schweckendiek, W. Treatment of psoriasis vulgaris. Med. Monatsschr 1959, 13, 103–104. [Google Scholar]
- Claussen, M.C.; Korn, T. Immune mechanisms of new therapeutic strategies in MS: Teriflunomide. Clin. Immunol 2012, 142, 49–56. [Google Scholar]
- Dimitrova, P.; Skapenko, A.; Herrmann, M.L.; Schleyerbach, R.; Kalden, J.R.; Schulze-Koops, H. Restriction of de novo pyrimidine biosynthesis inhibits Th1 cell activation and promotes Th2 cell differentiation. J. Immunol 2002, 169, 3392–3399. [Google Scholar]
- Xu, X.; Williams, J.W.; Bremer, E.G.; Finnegan, A.; Chong, A.S. Inhibition of protein tyrosine phosphorylation in T cells by a novel immunosuppressive agent, leflunomide. J. Biol. Chem 1995, 270, 12398–12403. [Google Scholar]
- Zeyda, M.; Poglitsch, M.; Geyeregger, R.; Smolen, J.S.; Zlabinger, G.J.; Horl, W.H.; Waldhausl, W.; Stulnig, T.M.; Saemann, M.D. Disruption of the interaction of T cells with antigen-presenting cells by the active leflunomide metabolite teriflunomide: Involvement of impaired integrin activation and immunologic synapse formation. Arthritis Rheum 2005, 52, 2730–2739. [Google Scholar]
- Korn, T.; Magnus, T.; Toyka, K.; Jung, S. Modulation of effector cell functions in experimental autoimmune encephalomyelitis by leflunomide—Mechanisms independent of pyrimidine depletion. J. Leukoc. Biol 2004, 76, 950–960. [Google Scholar]
- Merrill, J.E.; Hanak, S.; Pu, S.F.; Liang, J.; Dang, C.; Iglesias-Bregna, D.; Harvey, B.; Zhu, B.; McMonagle-Strucko, K. Teriflunomide reduces behavioral, electrophysiological, and histopathological deficits in the Dark Agouti rat model of experimental autoimmune encephalomyelitis. J. Neurol 2009, 256, 89–103. [Google Scholar]
- Wolinsky, J.S.; Narayana, P.A.; Noseworthy, J.H.; Lublin, F.D.; Whitaker, J.N.; Linde, A.; Gjorstrup, P.; Sullivan, H.C. North American Linomide Investigators. Linomide in relapsing and secondary progressive MS: Part II: MRI results. MRI Analysis Center of the University of Texas-Houston, Health Science Center, and the. Neurology 2000, 54, 1734–1741. [Google Scholar]
- Noseworthy, J.H.; Wolinsky, J.S.; Lublin, F.D.; Whitaker, J.N.; Linde, A.; Gjorstrup, P.; Sullivan, H.C. North American Linomide Investigators. Linomide in relapsing and secondary progressive MS: Part I: Trial design and clinical results. Neurology 2000, 54, 1726–1733. [Google Scholar]
- Tan, I.L.; Nijeholt, G.J.L.; Polman, C.H.; Ader, H.J.; Barkhof, F. Linomide in the treatment of multiple sclerosis: MRI results from prematurely terminated phase-III trials. Mult. Scler 2000, 6, 99–104. [Google Scholar]
- Wegner, C.; Stadelmann, C.; Pfortner, R.; Raymond, E.; Feigelson, S.; Alon, R.; Timan, B.; Hayardeny, L.; Bruck, W. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol 2010, 227, 133–143. [Google Scholar]
- Brunmark, C.; Runstrom, A.; Ohlsson, L.; Sparre, B.; Brodin, T.; Astrom, M.; Hedlund, G. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J. Neuroimmunol 2002, 130, 163–172. [Google Scholar]
- Yang, J.S.; Xu, L.Y.; Xiao, B.G.; Hedlund, G.; Link, H. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-beta in Lewis rats. J. Neuroimmunol 2004, 156, 3–9. [Google Scholar]
- Bruck, W.; Wegner, C. Insight into the mechanism of laquinimod action. J. Neurol. Sci 2011, 306, 173–179. [Google Scholar]
- Zou, L.P.; Abbas, N.; Volkmann, I.; Nennesmo, I.; Levi, M.; Wahren, B.; Winblad, B.; Hedlund, G.; Zhu, J. Suppression of experimental autoimmune neuritis by ABR-215062 is associated with altered Th1/Th2 balance and inhibited migration of inflammatory cells into the peripheral nerve tissue. Neuropharmacology 2002, 42, 731–739. [Google Scholar]
- Gurevich, M.; Gritzman, T.; Orbach, R.; Tuller, T.; Feldman, A.; Achiron, A. Laquinimod suppress antigen presentation in relapsing-remitting multiple sclerosis: In-vitro high-throughput gene expression study. J. Neuroimmunol 2010, 221, 87–94. [Google Scholar]
- Thone, J.; Ellrichmann, G.; Seubert, S.; Peruga, I.; Lee, D.H.; Conrad, R.; Hayardeny, L.; Comi, G.; Wiese, S.; Linker, R.A.; et al. Modulation of autoimmune demyelination by laquinimod via induction of brain-derived neurotrophic factor. Am. J. Pathol 2012, 180, 267–274. [Google Scholar]
- Linker, R.A.; Lee, D.H.; Demir, S.; Wiese, S.; Kruse, N.; Siglienti, I.; Gerhardt, E.; Neumann, H.; Sendtner, M.; Luhder, F.; et al. Functional role of brain-derived neurotrophic factor in neuroprotective autoimmunity: Therapeutic implications in a model of multiple sclerosis. Brain 2010, 133, 2248–2263. [Google Scholar]
- Lee, D.H.; Geyer, E.; Flach, A.C.; Jung, K.; Gold, R.; Flugel, A.; Linker, R.A.; Luhder, F. Central nervous system rather than immune cell-derived BDNF mediates axonal protective effects early in autoimmune demyelination. Acta Neuropathol 2012, 123, 247–258. [Google Scholar]
- Gilleece, M.H.; Dexter, T.M. Effect of Campath-1H antibody on human hematopoietic progenitors in vitro. Blood 1993, 82, 807–812. [Google Scholar]
- Coles, A.J.; Fox, E.; Vladic, A.; Gazda, S.K.; Brinar, V.; Selmaj, K.W.; Skoromets, A.; Stolyarov, I.; Bass, A.; Sullivan, H.; et al. Alemtuzumab more effective than interferon beta-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology 2012, 78, 1069–1078. [Google Scholar]
- Coles, A.J.; Cox, A.; Le Page, E.; Jones, J.; Trip, S.A.; Deans, J.; Seaman, S.; Miller, D.H.; Hale, G.; Waldmann, H.; et al. The window of therapeutic opportunity in multiple sclerosis: Evidence from monoclonal antibody therapy. J. Neurol 2006, 253, 98–108. [Google Scholar]
- Thompson, S.A.; Jones, J.L.; Cox, A.L.; Compston, D.A.; Coles, A.J. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J. Clin. Immunol 2010, 30, 99–105. [Google Scholar]
- Cox, A.L.; Thompson, S.A.; Jones, J.L.; Robertson, V.H.; Hale, G.; Waldmann, H.; Compston, D.A.; Coles, A.J. Lymphocyte homeostasis following therapeutic lymphocyte depletion in multiple sclerosis. Eur. J. Immunol 2005, 35, 3332–3342. [Google Scholar]
- Coles, A.J.; Compston, D.A.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Norris, K.; Tandon, P.K. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N. Engl. J. Med 2008, 359, 1786–1801. [Google Scholar]
- Jones, J.L.; Phuah, C.L.; Cox, A.L.; Thompson, S.A.; Ban, M.; Shawcross, J.; Walton, A.; Sawcer, S.J.; Compston, A.; Coles, A.J. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J. Clin. Invest 2009, 119, 2052–2061. [Google Scholar]
- Lutterotti, A.; Martin, R. Getting specific: Monoclonal antibodies in multiple sclerosis. Lancet Neurol 2008, 7, 538–547. [Google Scholar]
- Kim, H.P.; Imbert, J.; Leonard, W.J. Both integrated and differential regulation of components of the IL-2/IL-2 receptor system. Cytokine Growth Factor Rev 2006, 17, 349–366. [Google Scholar]
- Sen, H.N.; Levy-Clarke, G.; Faia, L.J.; Li, Z.; Yeh, S.; Barron, K.S.; Ryan, J.G.; Hammel, K.; Nussenblatt, R.B. High-dose daclizumab for the treatment of juvenile idiopathic arthritis-associated active anterior uveitis. Am. J. Ophthalmol 2009, 148. [Google Scholar]
- Yeh, S.; Wroblewski, K.; Buggage, R.; Li, Z.; Kurup, S.K.; Sen, H.N.; Dahr, S.; Sran, P.; Reed, G.F.; Robinson, R.; et al. High-dose humanized anti-IL-2 receptor alpha antibody (daclizumab) for the treatment of active, non-infectious uveitis. J. Autoimmun 2008, 31, 91–97. [Google Scholar]
- Buggage, R.R.; Levy-Clarke, G.; Sen, H.N.; Ursea, R.; Srivastava, S.K.; Suhler, E.B.; Altemare, C.; Velez, G.; Ragheb, J.; Chan, C.C.; et al. A double-masked, randomized study to investigate the safety and efficacy of daclizumab to treat the ocular complications related to Behcet’s disease. Ocul. Immunol. Inflamm 2007, 15, 63–70. [Google Scholar]
- Nussenblatt, R.B.; Fortin, E.; Schiffman, R.; Rizzo, L.; Smith, J.; van Veldhuisen, P.; Sran, P.; Yaffe, A.; Goldman, C.K.; Waldmann, T.A.; et al. Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: A phase I/II clinical trial. Proc. Natl. Acad. Sci. USA 1999, 96, 7462–7466. [Google Scholar]
- Webster, A.C.; Ruster, L.P.; McGee, R.; Matheson, S.L.; Higgins, G.Y.; Willis, N.S.; Chapman, J.R.; Craig, J.C. Interleukin 2 receptor antagonists for kidney transplant recipients. Cochrane Database Syst. Rev 2010, CD003897. [Google Scholar]
- Wynn, D.; Kaufman, M.; Montalban, X.; Vollmer, T.; Simon, J.; Elkins, J.; O’Neill, G.; Neyer, L.; Sheridan, J.; Wang, C.; et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): A phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol 2010, 9, 381–390. [Google Scholar]
- Giovannoni, G.; Gold, R.; Selmaj, K.; Havrdova, E.; Montalban, X.; Radue, E.W.; Stefoski, D.; Robinson, R.; Riester, K.; Elkins, J.; et al. A randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of daclizumab HYP monotherapy in relasping-remitting multiple sclerosis: Primary results of the SELECT trial. Proceedings of European and Americas Committees on Treatment and Research in Multiple Sclerosis (ECTRIMS), Amsterdam, The Netherlands, 19–22 October 2011.
- Martin, J.F.; Perry, J.S.; Jakhete, N.R.; Wang, X.; Bielekova, B. An IL-2 paradox: Blocking CD25 on T cells induces IL-2-driven activation of CD56(bright) NK cells. J. Immunol 2010, 185, 1311–1320. [Google Scholar]
- Hao, J.; Campagnolo, D.; Liu, R.; Piao, W.; Shi, S.; Hu, B.; Xiang, R.; Zhou, Q.; Vollmer, T.; van Kaer, L.; et al. Interleukin-2/interleukin-2 antibody therapy induces target organ natural killer cells that inhibit central nervous system inflammation. Ann. Neurol 2011, 69, 721–734. [Google Scholar]
- Bielekova, B.; Howard, T.; Packer, A.N.; Richert, N.; Blevins, G.; Ohayon, J.; Waldmann, T.A.; McFarland, H.F.; Martin, R. Effect of anti-CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch. Neurol 2009, 66, 483–489. [Google Scholar]
- Fehniger, T.A.; Bluman, E.M.; Porter, M.M.; Mrozek, E.; Cooper, M.A.; VanDeusen, J.B.; Frankel, S.R.; Stock, W.; Caligiuri, M.A. Potential mechanisms of human natural killer cell expansion in vivo during low-dose IL-2 therapy. J. Clin. Invest 2000, 106, 117–124. [Google Scholar]
- Hao, J.; Liu, R.; Piao, W.; Zhou, Q.; Vollmer, T.L.; Campagnolo, D.I.; Xiang, R.; La Cava, A.; van Kaer, L.; Shi, F.D. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J. Exp. Med 2010, 207, 1907–1921. [Google Scholar]
- Bielekova, B.; Catalfamo, M.; Reichert-Scrivner, S.; Packer, A.; Cerna, M.; Waldmann, T.A.; McFarland, H.; Henkart, P.A.; Martin, R. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5941–5946. [Google Scholar]
- Wuest, S.C.; Edwan, J.H.; Martin, J.F.; Han, S.; Perry, J.S.; Cartagena, C.M.; Matsuura, E.; Maric, D.; Waldmann, T.A.; Bielekova, B. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat. Med 2011, 17, 604–609. [Google Scholar]
- Snyder, J.T.; Shen, J.; Azmi, H.; Hou, J.; Fowler, D.H.; Ragheb, J.A. Direct inhibition of CD40L expression can contribute to the clinical efficacy of daclizumab independently of its effects on cell division and Th1/Th2 cytokine production. Blood 2007, 109, 5399–5406. [Google Scholar]
- Barun, B.; Bar-Or, A. Treatment of multiple sclerosis with anti-CD20 antibodies. Clin. Immunol 2012, 142, 31–37. [Google Scholar]
- Edwards, J.C.; Cambridge, G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat. Rev. Immunol 2006, 6, 394–403. [Google Scholar]
- Clifford, D.B.; Ances, B.; Costello, C.; Rosen-Schmidt, S.; Andersson, M.; Parks, D.; Perry, A.; Yerra, R.; Schmidt, R.; Alvarez, E.; et al. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch. Neurol 2011, 68, 1156–1164. [Google Scholar]
- Molloy, E.S.; Calabrese, L.H. Progressive multifocal leukoencephalopathy associated with immunosuppressive therapy in rheumatic diseases: Evolving role of biologic therapies. Arthritis Rheum 2012, 64, 3043–3051. [Google Scholar]
- Tuccori, M.; Focosi, D.; Maggi, F.; Cosottini, M.; Meini, B.; Lena, F.; Blandizzi, C.; del Tacca, M.; Petrini, M. Progressive multifocal leukoencephalopathy: A report of three cases in HIV-negative patients with non-Hodgkin’s lymphomas treated with rituximab. Ann. Hematol 2010, 89, 519–522. [Google Scholar]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol 2009, 66, 460–471. [Google Scholar]
- He, D.; Zhou, H.; Han, W.; Zhang, S. Rituximab for relapsing-remitting multiple sclerosis. Cochrane Database Syst. Rev 2011. [Google Scholar] [CrossRef]
- Piccio, L.; Naismith, R.T.; Trinkaus, K.; Klein, R.S.; Parks, B.J.; Lyons, J.A.; Cross, A.H. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch. Neurol 2010, 67, 707–714. [Google Scholar]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J. Exp. Med 2012, 209, 1001–1010. [Google Scholar]
- Genovese, M.C.; Kaine, J.L.; Lowenstein, M.B.; Del Giudice, J.; Baldassare, A.; Schechtman, J.; Fudman, E.; Kohen, M.; Gujrathi, S.; Trapp, R.G.; et al. Ocrelizumab, a humanized anti-CD20 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I/II randomized, blinded, placebo-controlled, dose-ranging study. Arthritis Rheum 2008, 58, 2652–2661. [Google Scholar]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; Hauser, S.L. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar]
- Stohl, W.; Gomez-Reino, J.; Olech, E.; Dudler, J.; Fleischmann, R.M.; Zerbini, C.A.; Ashrafzadeh, A.; Grzeschik, S.; Bieraugel, R.; Green, J.; et al. Safety and efficacy of ocrelizumab in combination with methotrexate in MTX-naive subjects with rheumatoid arthritis: The phase III FILM trial. Ann. Rheum. Dis 2012. [Google Scholar] [CrossRef]
- Harigai, M.; Tanaka, Y.; Maisawa, S. Safety and efficacy of various dosages of ocrelizumab in Japanese patients with rheumatoid arthritis with an inadequate response to methotrexate therapy: A placebo-controlled double-blind parallel-group study. J. Rheumatol 2012, 39, 486–495. [Google Scholar]
- Hartung, H.P.; Kieseier, B.C. Atacicept: Targeting B cells in multiple sclerosis. Ther. Adv. Neurol. Disord 2010, 3, 205–216. [Google Scholar]
- Dillon, S.R.; Gross, J.A.; Ansell, S.M.; Novak, A.J. An APRIL to remember: Novel TNF ligands as therapeutic targets. Nat. Rev. Drug Discov 2006, 5, 235–246. [Google Scholar]
- Thangarajh, M.; Gomes, A.; Masterman, T.; Hillert, J.; Hjelmstrom, P. Expression of B-cell-activating factor of the TNF family (BAFF) and its receptors in multiple sclerosis. J. Neuroimmunol 2004, 152, 183–190. [Google Scholar]
- Thangarajh, M.; Masterman, T.; Hillert, J.; Moerk, S.; Jonsson, R. A proliferation-inducing ligand (APRIL) is expressed by astrocytes and is increased in multiple sclerosis. Scand. J. Immunol 2007, 65, 92–98. [Google Scholar]
- Thangarajh, M.; Masterman, T.; Rot, U.; Duvefelt, K.; Brynedal, B.; Karrenbauer, V.D.; Hillert, J. Increased levels of APRIL (a proliferation-inducing ligand) mRNA in multiple sclerosis. J. Neuroimmunol 2005, 167, 210–214. [Google Scholar]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med 1999, 190, 1697–1710. [Google Scholar]
- Gross, J.A.; Dillon, S.R.; Mudri, S.; Johnston, J.; Littau, A.; Roque, R.; Rixon, M.; Schou, O.; Foley, K.P.; Haugen, H.; et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease. Impaired B cell maturation in mice lacking BLyS. Immunity 2001, 15, 289–302. [Google Scholar]
- Carbonatto, M.; Yu, P.; Bertolino, M.; Vigna, E.; Steidler, S.; Fava, L.; Daghero, C.; Roattino, B.; Onidi, M.; Ardizzone, M.; et al. Nonclinical safety, pharmacokinetics, and pharmacodynamics of atacicept. Toxicol. Sci 2008, 105, 200–210. [Google Scholar]
- Ginzler, E.M.; Wax, S.; Rajeswaran, A.; Copt, S.; Hillson, J.; Ramos, E.; Singer, N.G. Atacicept in combination with MMF and corticosteroids in lupus nephritis: Results of a prematurely terminated trial. Arthritis Res. Ther 2012, 14, R33. [Google Scholar]
- Van Vollenhoven, R.F.; Kinnman, N.; Vincent, E.; Wax, S.; Bathon, J. Atacicept in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a phase II, randomized, placebo-controlled trial. Arthritis Rheum 2011, 63, 1782–1792. [Google Scholar]
- Tak, P.P.; Thurlings, R.M.; Rossier, C.; Nestorov, I.; Dimic, A.; Mircetic, V.; Rischmueller, M.; Nasonov, E.; Shmidt, E.; Emery, P.; Munafo, A. Atacicept in patients with rheumatoid arthritis: Results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating, single- and repeated-dose study. Arthritis Rheum 2008, 58, 61–72. [Google Scholar]
- Harp, C.T.; Ireland, S.; Davis, L.S.; Remington, G.; Cassidy, B.; Cravens, P.D.; Stuve, O.; Lovett-Racke, A.E.; Eagar, T.N.; Greenberg, B.M.; et al. Memory B cells from a subset of treatment-naive relapsing-remitting multiple sclerosis patients elicit CD4(+) T-cell proliferation and IFN-gamma production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur. J. Immunol 2010, 40, 2942–2956. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Agent [ref] | Mechanism of action | Route of admin | Efficacy | Adverse effects | ||||
|---|---|---|---|---|---|---|---|---|
| ARR | Prog | MRI | Nature | Level | Freq | |||
| Currently approved therapies | ||||||||
| β-interferon † [14,91–94] | Promotes Th2 environment Anti-inflammatory (reduced IL-17; IL-23) NK; T-reg expansion | SC/IM | + | (+) | ++ | Injection site reactions | + | ++ |
| Flu-like symptoms | + | ++ | ||||||
| Depression | + | ++ | ||||||
| Lymphopaenia | + | +++ | ||||||
| LFT abnormality | + | ++ | ||||||
| Neutralising antibodies | (+) | ++ | ||||||
| Glatiramer acetate [95] | Promotes Th2 environment Modulation of APCs Reduced TNF; IL-12 | SC | + | − | + | Injection site reaction | + | +++# |
| Post-injection reaction | + | +++ | ||||||
| Natalizumab [96] | Antibody blockade of VLA-4 | IV | +++ | + | +++ | Infusion reaction | ++ | ++ |
| PML | +++ | + | ||||||
| Neutralising antibodies | + | + | ||||||
| Fingolimod [97,98] | Blockade of S-1-P1 receptors Trapping of lymphocytes in lymph node (Neuroprotection) | Oral | ++ | + | +++ | Bradycardia | ++ | ++ |
| Macular oedema | ++ | + | ||||||
| LFT abnormality | + | +++ | ||||||
| Lymphopaenia | ++ | ++ | ||||||
| Emerging therapies | ||||||||
| Dimethyl fumarate [99–101] | Reduces oxidative stress Increased Nrf2 | Oral | ++ | + | +++ | Gastrointestinal symptoms | ++ | +++ |
| Flushing | + | +++ | ||||||
| Teriflunomide [102] | Dihydroorotate dehydrogenase inhibitor Prevents pyrimidine synthesis Restricts lymphocyte proliferation | Oral | + | + | ++ | Paraesthesia | + | +++ |
| LFT abnormality | + | +++ | ||||||
| Gastrointestinal symptoms | + | +++ | ||||||
| Arthralgia | + | +++ | ||||||
| Alopecia | + | +++ | ||||||
| Laquinimod [103] | Quinolone-3-carboxamide Promotes Th2 shift (Neuroprotection) | Oral | + | ++ | + | LFT abnormality | + | +++ |
| Arthralgia | + | ++ | ||||||
| Back pain | + | +++ | ||||||
| Alemtuzumab [104,105] | CD52 monoclonal antibody Lysis of lymphocytes | IV | +++ | + | +++ | Thyroid autoimmunity | ++ | ++ |
| ITP | ++ | + | ||||||
| Goodpasture’s syndrome | +++ | + | ||||||
| Oral/genital herpes * | + | ++ | ||||||
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Buzzard, K.A.; Broadley, S.A.; Butzkueven, H. What Do Effective Treatments for Multiple Sclerosis Tell Us about the Molecular Mechanisms Involved in Pathogenesis? Int. J. Mol. Sci. 2012, 13, 12665-12709. https://doi.org/10.3390/ijms131012665
Buzzard KA, Broadley SA, Butzkueven H. What Do Effective Treatments for Multiple Sclerosis Tell Us about the Molecular Mechanisms Involved in Pathogenesis? International Journal of Molecular Sciences. 2012; 13(10):12665-12709. https://doi.org/10.3390/ijms131012665
Chicago/Turabian StyleBuzzard, Katherine A., Simon A. Broadley, and Helmut Butzkueven. 2012. "What Do Effective Treatments for Multiple Sclerosis Tell Us about the Molecular Mechanisms Involved in Pathogenesis?" International Journal of Molecular Sciences 13, no. 10: 12665-12709. https://doi.org/10.3390/ijms131012665