Fragment-Based Screening by Protein Crystallography: Successes and Pitfalls


Abstract
:1. Introduction
2. Practical Considerations in FBDD
2.1. Library and Compound Properties
2.2. The Target
2.3. X-ray Sources, Detectors and Robots and Software
2.4. Potential Pitfalls in X-ray Based Screening
3. Examples of Crystallography in FBDD
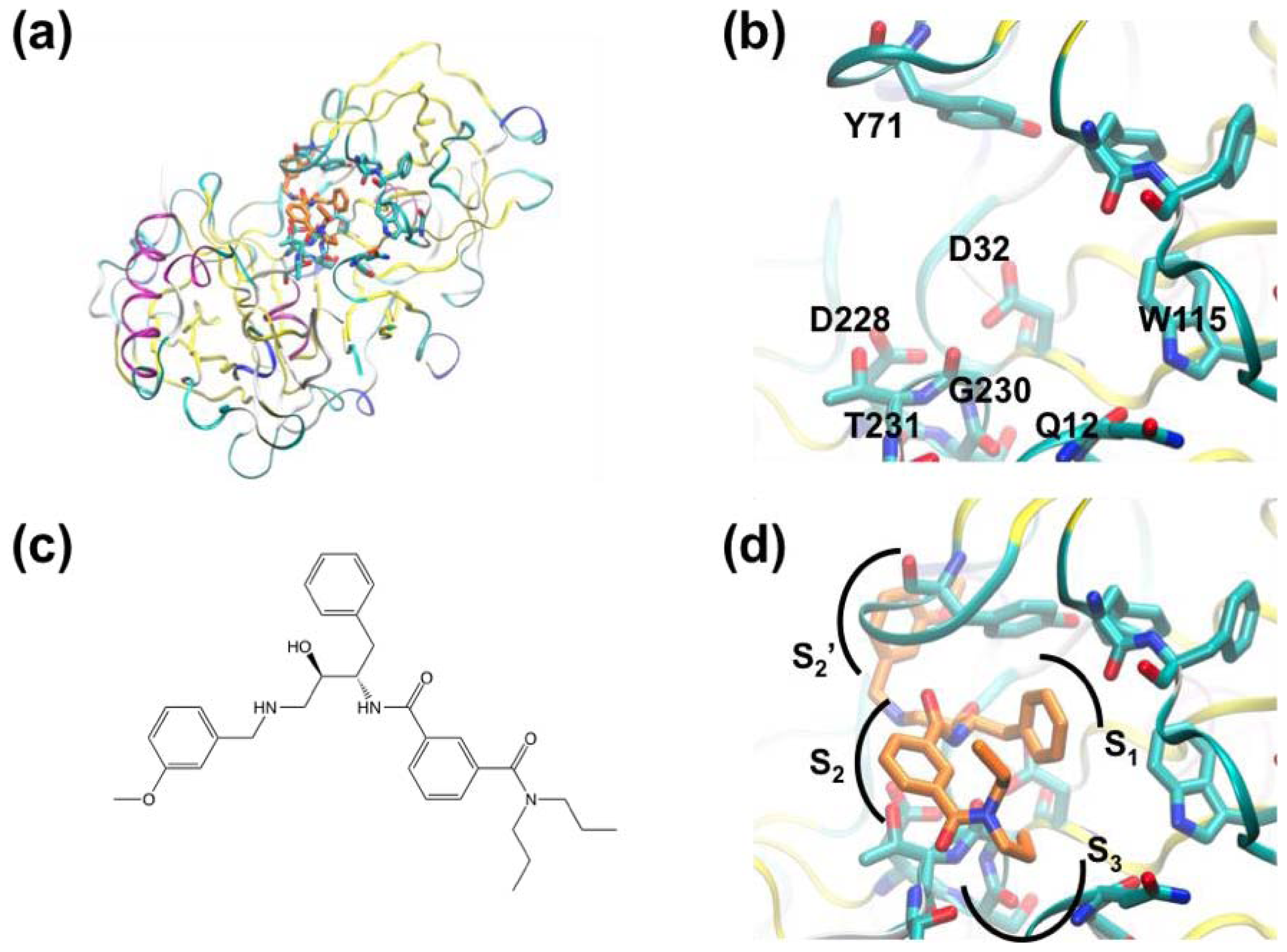
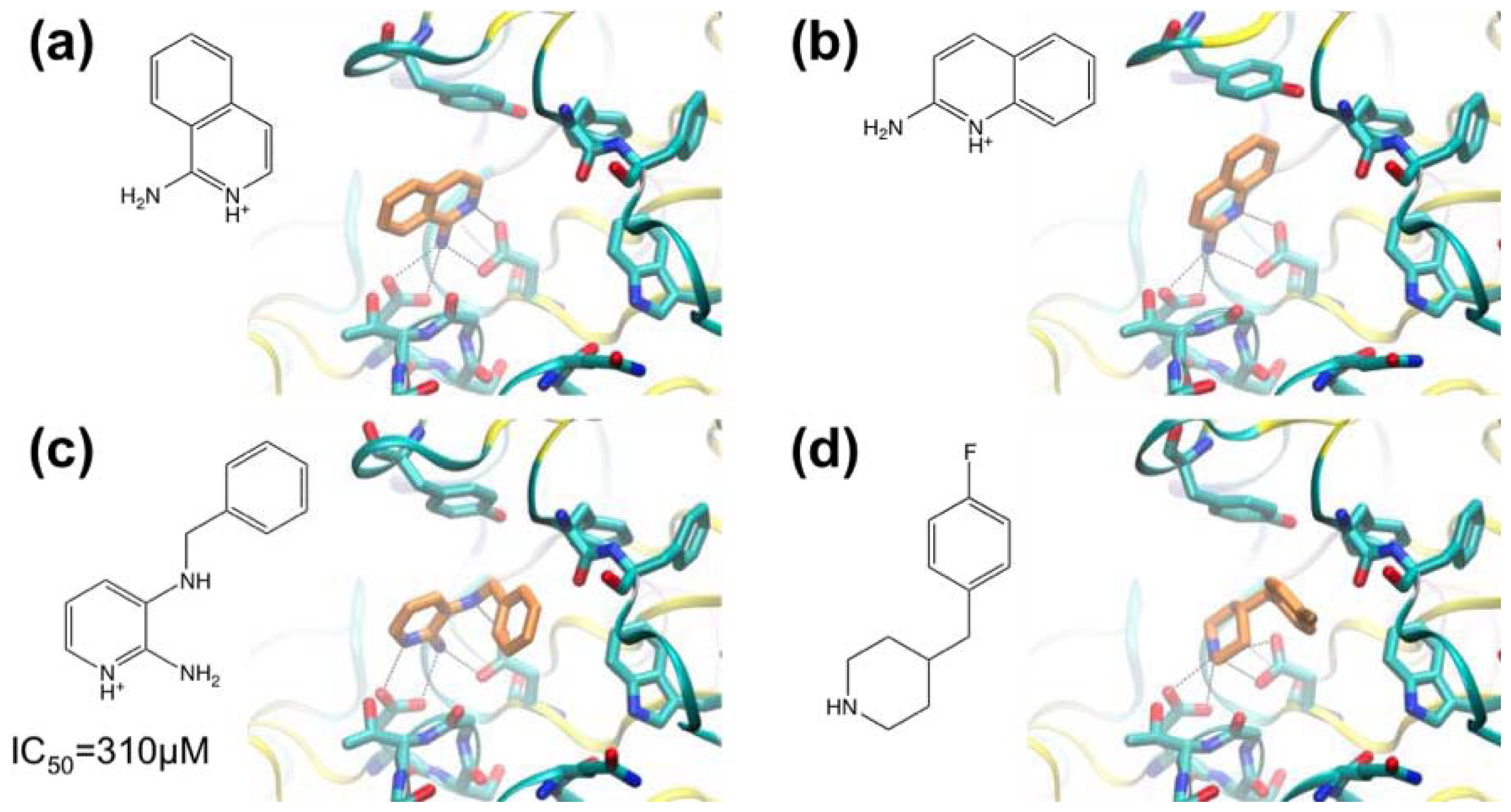
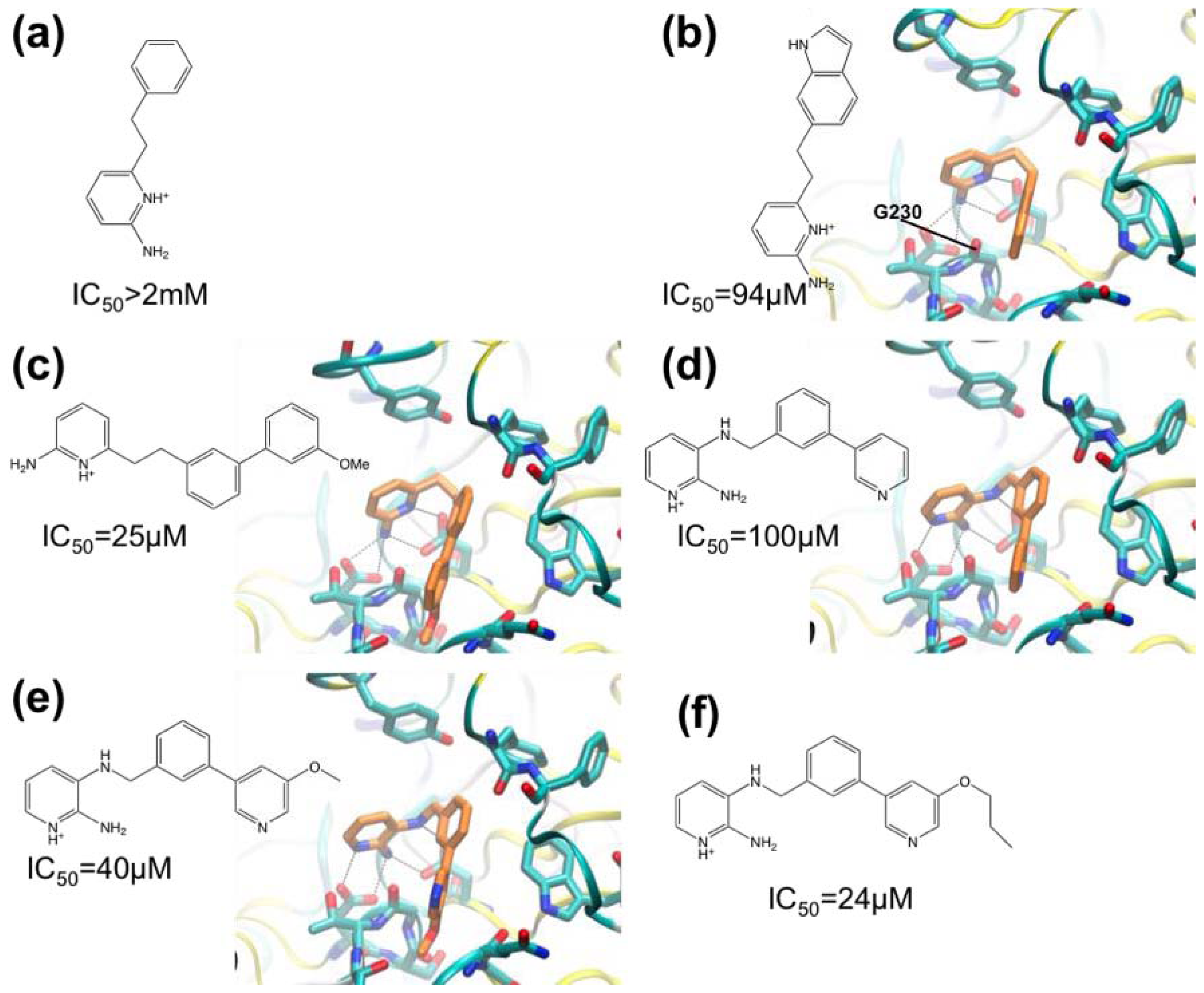
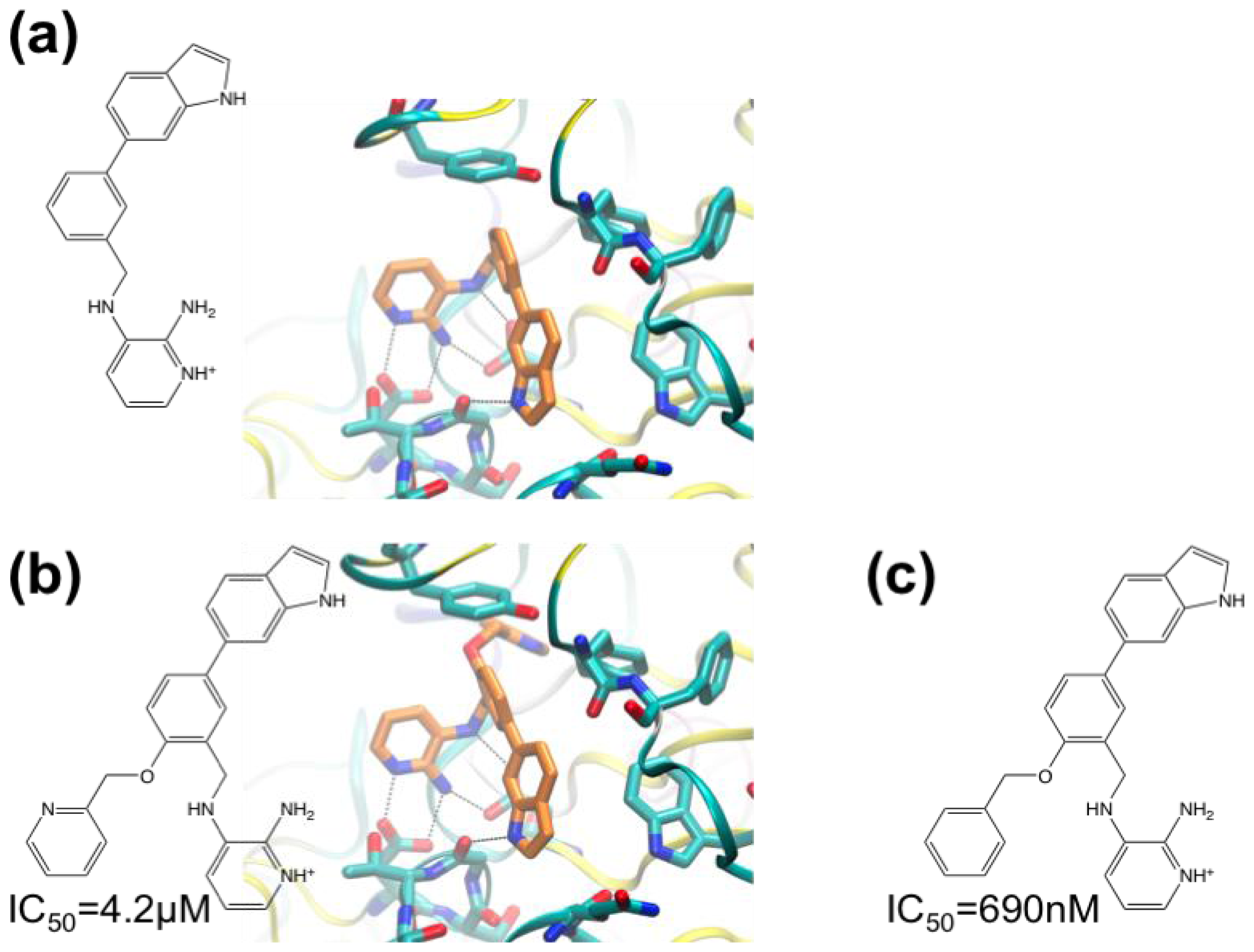
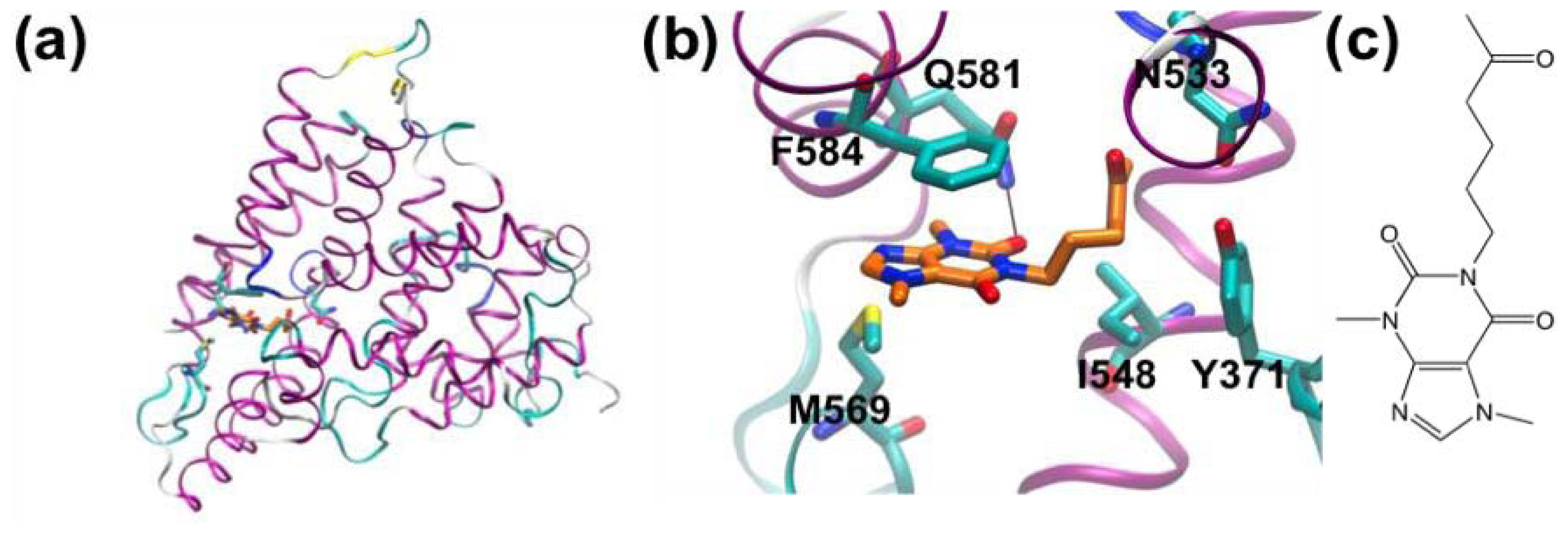
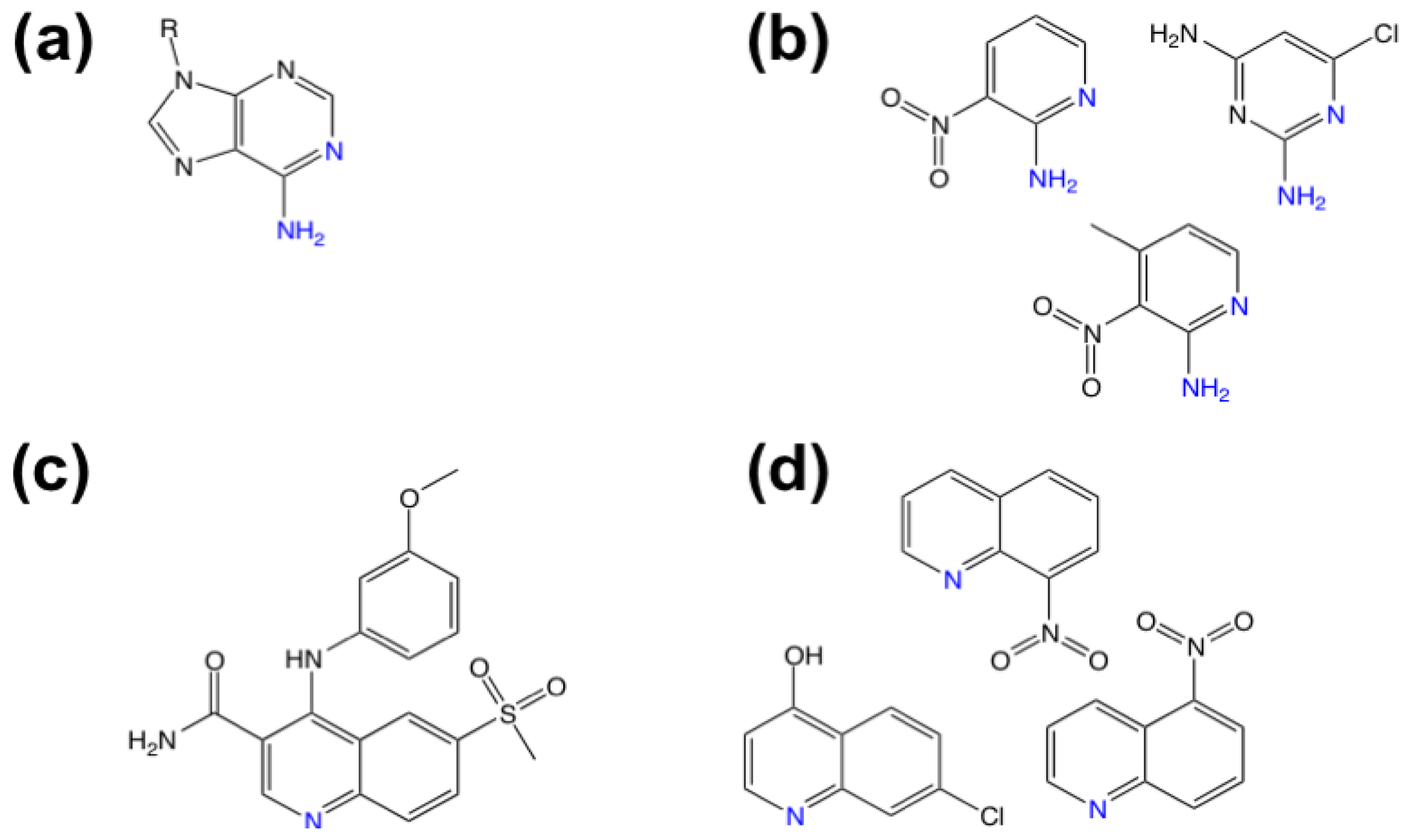
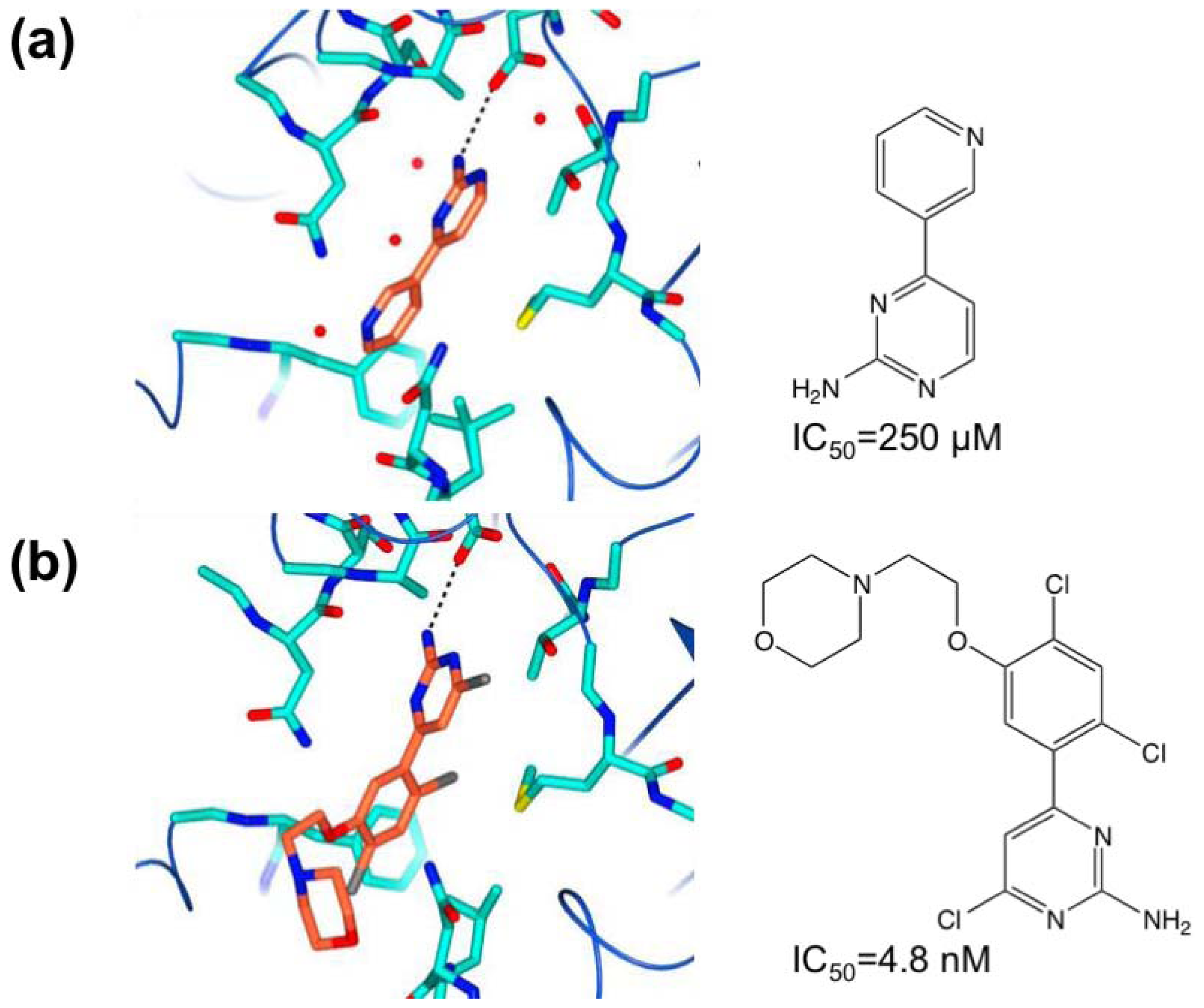
3.1. β-Secretase
3.2. Phosphodiesterase 4A
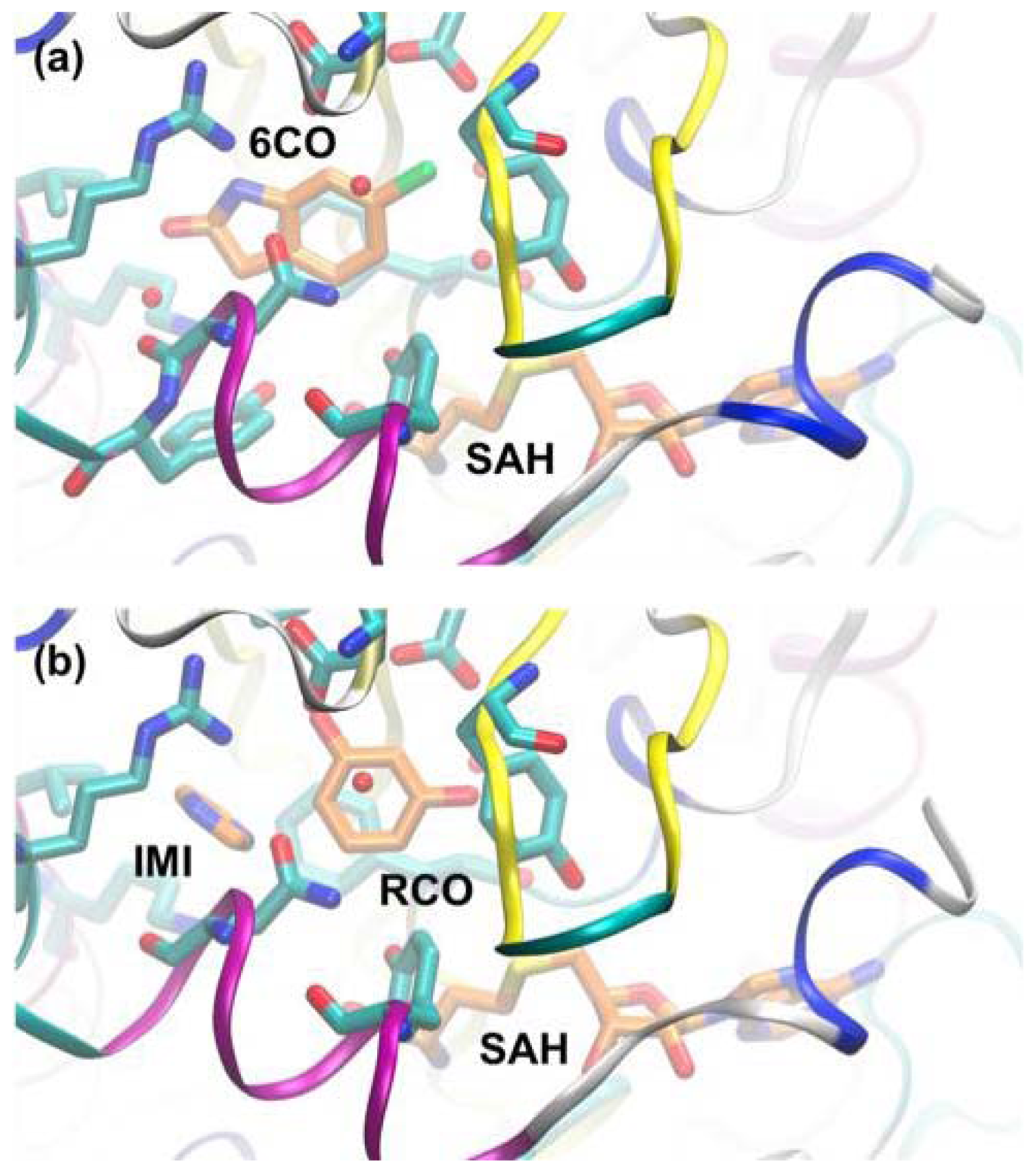
3.3. A Case of Cooperative Fragment Binding: hPNMT
3.4. Heat Shock Protein 90 (Hsp90)
4. Conclusions
Acknowledgment
- Conflict of InterestThe authors declare no conflict of interest.
References
- Larsson, A.; Jansson, A.; Aberg, A.; Nordlund, P. Efficiency of hit generation and structural characterization in fragment-based ligand discovery. Curr. Opin. Chem. Biol 2012, 15, 482–488. [Google Scholar]
- Davies, T.G.; Tickle, I.J. Fragment screening using X-ray crystallography. Top. Curr. Chem 2012, 317, 33–59. [Google Scholar]
- Fitzpatrick, P.A.; Steinmetz, A.C.; Ringe, D.; Klibanov, A.M. Enzyme crystal structure in a neat organic solvent. Proc. Natl. Acad. Sci. USA 1993, 90, 8653–8657. [Google Scholar]
- English, A.C.; Done, S.H.; Caves, L.S.; Groom, C.R.; Hubbard, R.E. Locating interaction sites on proteins: The crystal structure of thermolysin soaked in 2% to 100% isopropanol. Proteins 1999, 37, 628–640. [Google Scholar]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar]
- Stout, T.J.; Sage, C.R.; Stroud, R.M. The additivity of substrate fragments in enzyme-ligand binding. Structure 1998, 6, 839–848. [Google Scholar]
- Lesuisse, D.; Lange, G.; Deprez, P.; Benard, D.; Schoot, B.; Delettre, G.; Marquette, J.P.; Broto, P.; Jean-Baptiste, V.; Bichet, P.; et al. SAR and X-ray. A new approach combining fragment-based screening and rational drug design: Application to the discovery of nanomolar inhibitors of Src SH2. J. Med. Chem 2002, 45, 2379–2387. [Google Scholar]
- Maly, D.J.; Choong, I.C.; Ellman, J.A. Combinatorial target-guided ligand assembly: Identification of potent subtype-selective c-Src inhibitors. Proc. Natl. Acad. Sci. USA 2000, 97, 2419–2424. [Google Scholar]
- Boehm, H.J.; Boehringer, M.; Bur, D.; Gmuender, H.; Huber, W.; Klaus, W.; Kostrewa, D.; Kuehne, H.; Luebbers, T.; Meunier-Keller, N.; et al. Novel inhibitors of DNA gyrase: 3D structure based biased needle screening, hit validation by biophysical methods, and 3D guided optimization. A promising alternative to random screening. J. Med. Chem 2000, 43, 2664–2674. [Google Scholar]
- Fejzo, J.; Lepre, C.A.; Peng, J.W.; Bemis, G.W.; Ajay Murcko, M.A.; Moore, J.M. The SHAPES strategy: An NMR-based approach for lead generation in drug discovery. Chem. Biol. 1999, 6, 755–769. [Google Scholar]
- Liebeschuetz, J.W.; Jones, S.D.; Morgan, P.J.; Murray, C.W.; Rimmer, A.D.; Roscoe, J.M.; Waszkowycz, B.; Welsh, P.M.; Wylie, W.A.; Young, S.C.; et al. PRO_SELECT: Combining structure-based drug design and array-based chemistry for rapid lead discovery. 2. The development of a series of highly potent and selective factor Xa inhibitors. J. Med. Chem 2002, 45, 1221–1232. [Google Scholar]
- Wyss, D.F.; Wang, Y.S.; Eaton, H.L.; Strickland, C.; Voigt, J.H.; Zhu, Z.; Stamford, A.W. Combining NMR and X-ray crystallography in fragment-based drug discovery: Discovery of highly potent and selective BACE-1 inhibitors. Top. Curr. Chem 2012, 317, 83–114. [Google Scholar]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci 2001, 41, 856–864. [Google Scholar]
- Murray, C.W.; Verdonk, M.L. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J. Comput. Aided. Mol. Des 2002, 16, 741–753. [Google Scholar]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar]
- Carr, R.A.E.; Congreve, M.; Murray, C.W.; Rees, D.C. Fragment-based lead discovery: Leads by design. Drug Discov. Today 2005, 10, 987–992. [Google Scholar]
- Ciulli, A.; Abell, C. Fragment-based approaches to enzyme inhibition. Curr. Opin. Biotechnol 2007, 18, 489–496. [Google Scholar]
- Kranz, J.K.; Schalk-Hihi, C. Protein thermal shifts to identify low molecular weight fragments. Methods Enzymol 2011, 493, 277–298. [Google Scholar]
- Giannetti, A.M. From experimental design to validated hits a comprehensive walk-through of fragment lead identification using surface plasmon resonance. Methods Enzymol 2011, 493, 169–218. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev 2001, 46, 3–26. [Google Scholar]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar]
- Hung, A.W.; Ramek, A.; Wang, Y.; Kaya, T.; Wilson, J.A.; Clemons, P.A.; Young, D.W. Route to three-dimensional fragments using diversity-oriented synthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 6799–6804. [Google Scholar]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev 1996, 16, 3–50. [Google Scholar]
- Drew, K.L.M.; Baiman, H.; Khwaounjoo, P.; Yu, B.; Reynisson, J. Size estimation of chemical space: How big is it? J. Pharm. Pharmacol 2012, 64, 490–495. [Google Scholar]
- Fink, T.; Reymond, J.L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: Assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model 2007, 47, 342–353. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci 1988, 28, 31–36. [Google Scholar]
- Kong, F.; Yuan, L.; Zheng, Y.F.; Chen, W. Automatic liquid handling for life science: A critical review of the current state of the art. J. Lab. Autom 2012, 17, 169–185. [Google Scholar]
- Chayen, N.E. Optimization Techniques for Automation and High Throughput. In Methods in Molecular Biology; Doublié, S., Ed.; Humana Press: Totowa, NJ, USA, 2007; Volume 363, pp. 175–190. [Google Scholar]
- Grueninger-Leitch, F.; D’Arcy, A.; D’Arcy, B.; Chene, C. Deglycosylation of proteins for crystallization using recombinant fusion protein glycosidases. Protein Sci 1996, 5, 2617–2622. [Google Scholar]
- Dong, A.; Xu, X.; Edwards, A.M.; Chang, C.; Chruszcz, M.; Cuff, M.; Cymborowski, M.; Di Leo, R.; Egorova, O.; Evdokimova, E.; et al. In situ proteolysis for protein crystallization and structure determination. Nat. Methods 2007, 4, 1019–1021. [Google Scholar]
- Derewenda, Z.S. Application of protein engineering to enhance crystallizability and improve crystal properties. Acta Crystallogr. D 2010, 66, 604–615. [Google Scholar]
- Jenkins, T.M.; Hickman, A.B.; Dyda, F.; Ghirlando, R.; Davies, D.R.; Craigie, R. Catalytic domain of human immunodeficiency virus type 1 integrase: Identification of a soluble mutant by systematic replacement of hydrophobic residues. Proc. Natl. Acad. Sci. USA 1995, 92, 6057–6061. [Google Scholar]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [Google Scholar]
- Zhou, H.; Brock, J.; Liu, D.; Board, P.G.; Oakley, A.J. Structural insights into the dehydroascorbate reductase activity of human omega-class glutathione transferases. J. Mol. Biol 2012, 420, 190–203. [Google Scholar]
- Patel, S.B.; Cameron, P.M.; Frantz-Wattley, B.; O’Neill, E.; Becker, J.W.; Scapin, G. Lattice stabilization and enhanced diffraction in human p38α crystals by protein engineering. Biochim. Biophys. Acta 2004, 1696, 67–73. [Google Scholar]
- Badger, J. Crystallographic fragment screening. Methods Mol. Biol 2012, 841, 161–177. [Google Scholar]
- Wasserman, S.R.; Koss, J.W.; Sojitra, S.T.; Morisco, L.L.; Burley, S.K. Rapid-access, high-throughput synchrotron crystallography for drug discovery. Trends Pharmacol. Sci 2012, 33, 261–267. [Google Scholar]
- Hülsen, G.; Broennimann, C.; Eikenberry, E.F.; Wagner, A. Protein crystallography with a novel large-area pixel detector. J. Appl. Crystallogr 2006, 39, 550–557. [Google Scholar]Int. J. Mol. Sci 2012, 13, 12877.
- De Sanctis, D.; Beteva, A.; Caserotto, H.; Dobias, F.; Gabadinho, J.; Giraud, T.; Gobbo, A.; Guijarro, M.; Lentini, M.; Lavault, B.; et al. ID29: A high-intensity highly automated ESRF beamline for macromolecular crystallography experiments exploiting anomalous scattering. J. Synchrotron. Radiat 2012, 19, 455–461. [Google Scholar]
- Smith, C.A.; Cohen, A.E. The stanford automated mounter: Enabling high-throughput protein crystal screening at SSRL. J. Lab. Autom 2008, 13, 335–343. [Google Scholar]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron. Radiat 2002, 9, 401–406. [Google Scholar]
- Beteva, A.; Cipriani, F.; Cusack, S.; Delageniere, S.; Gabadinho, J.; Gordon, E.J.; Guijarro, M.; Hall, D.R.; Larsen, S.; Launer, L.; et al. High-throughput sample handling and data collection at synchrotrons: Embedding the ESRF into the high-throughput gene-to-structure pipeline. Acta Crystallogr. D 2006, 62, 1162–1169. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar]
- Terwilliger, T.C.; Adams, P.D.; Moriarty, N.W.; Cohn, J.D. Ligand identification using electron-density map correlations. Acta Crystallogr. D 2007, 63, 101–107. [Google Scholar]
- Evrard, G.X.; Langer, G.G.; Perrakis, A.; Lamzin, V.S. Assessment of automatic ligand building in ARP/wARP. Acta Crystallogr. D 2007, 63, 108–117. [Google Scholar]
- Mooij, W.T.; Hartshorn, M.J.; Tickle, I.J.; Sharff, A.J.; Verdonk, M.L.; Jhoti, H. Automated protein-ligand crystallography for structure-based drug design. ChemMedChem 2006, 1, 827–838. [Google Scholar]
- Jones, T.A.; Kjeldgaard, M. Electron-density map interpretation. Methods Enzymol 1997, 277, 173–208. [Google Scholar]
- Davis, A.M.; Teague, S.J.; Kleywegt, G.J. Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew. Chem. Int. Ed. Engl 2003, 42, 2718–2736. [Google Scholar]
- Davis, A.M.; St-Gallay, S.A.; Kleywegt, G.J. Limitations and lessons in the use of X-ray structural information in drug design. Drug Discov. Today 2008, 13, 831–841. [Google Scholar]
- Malde, A.K.; Mark, A.E. Challenges in the determination of the binding modes of non-standard ligands in X-ray crystal complexes. J. Comput. Aided Mol. Des 2011, 25, 1–12. [Google Scholar]
- Kleywegt, G.J.; Henrick, K.; Dodson, E.J.; van Aalten, D.M. Pound-wise but penny-foolish: How well do micromolecules fare in macromolecular refinement? Structure 2003, 11, 1051–1059. [Google Scholar]
- Ward, R.A.; Brassington, C.; Breeze, A.L.; Caputo, A.; Critchlow, S.; Davies, G.; Goodwin, L.; Hassall, G.; Greenwood, R.; Holdgate, G.A.; et al. Design and synthesis of novel lactate dehydrogenase a inhibitors by fragment-based lead generation. J. Med. Chem 2012, 55, 3285–3306. [Google Scholar]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar]
- Murray, C.W.; Callaghan, O.; Chessari, G.; Cleasby, A.; Congreve, M.; Frederickson, M.; Hartshorn, M.J.; McMenamin, R.; Patel, S.; Wallis, N. Application of fragment screening by X-ray crystallography to beta-secretase. J. Med. Chem 2007, 50, 1116–1123. [Google Scholar]
- Congreve, M.; Aharony, D.; Albert, J.; Callaghan, O.; Campbell, J.; Carr, R.A.E.; Chessari, G.; Cowan, S.; Edwards, P.D.; Frederickson, M.; et al. Application of fragment screening by X-ray crystallography to the discovery of aminopyridines as inhibitors of beta-secretase. J. Med. Chem 2007, 50, 1124–1132. [Google Scholar]
- Nair, P.C.; Malde, A.K.; Drinkwater, N.; Mark, A.E. Missing fragments: Detecting cooperative binding in fragment-based drug design. ACS Med. Chem. Lett 2012, 3, 322–326. [Google Scholar]
- Recht, M.I.; Sridhar, V.; Badger, J.; Hernandez, L.; Chie-Leon, B.; Nienaber, V.; Torres, F.E. Fragment-based screening for inhibitors of PDE4A using enthalpy arrays and X-ray crystallography. J. Biomol. Screen 2012, 17, 469–480. [Google Scholar]
- Murray, C.W.; Carr, M.G.; Callaghan, O.; Chessari, G.; Congreve, M.; Cowan, S.; Coyle, J.E.; Downham, R.; Figueroa, E.; Frederickson, M.; et al. Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency. J. Med. Chem 2010, 53, 5942–5955. [Google Scholar]
- Woodhead, A.J.; Angove, H.; Carr, M.G.; Chessari, G.; Congreve, M.; Coyle, J.E.; Cosme, J.; Graham, B.; Day, P.J.; Downham, R.; et al. Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5- (4-methylpiperazin-1-ylmethyl)-1,3-dihydrois oindol-2-yl]methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J. Med. Chem 2010, 53, 5956–5969. [Google Scholar]
- Chung, C.W.; Dean, A.W.; Woolven, J.M.; Bamborough, P. Fragment-based discovery of bromodomain inhibitors part 1: Inhibitor binding modes and implications for lead discovery. J. Med. Chem 2012, 55, 576–586. [Google Scholar]
- Bamborough, P.; Diallo, H.; Goodacre, J.D.; Gordon, L.; Lewis, A.; Seal, J.T.; Wilson, D.M.; Woodrow, M.D.; Chung, C.-W. Fragment-based discovery of bromodomain inhibitors part 2: Optimization of phenylisoxazole sulfonamides. J. Med. Chem 2011, 55, 587–596. [Google Scholar]
- Nichols, D.A.; Jaishankar, P.; Larson, W.; Smith, E.; Liu, G.; Beyrouthy, R.; Bonnet, R.; Renslo, A.R.; Chen, Y. Structure-based design of potent and ligand-efficient inhibitors of CTX-M Class A β-lactamase. J. Med. Chem 2012, 55, 2163–2172. [Google Scholar]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem 2012, 55, 1898–1903. [Google Scholar]
- Certal, V.; Halley, F.; Virone-Oddos, A.; Delorme, C.; Karlsson, A.; Rak, A.; Thompson, F.; Filoche-Rommé, B.; El-Ahmad, Y.; Carry, J.-C.; et al. Discovery and optimization of new benzimidazole- and benzoxazole-pyrimidone selective PI3Kβ inhibitors for the treatment of phosphatase and TENsin homologue (PTEN)-deficient cancers. J. Med. Chem 2012, 55, 4788–4805. [Google Scholar]
- Gitto, R.; Damiano, F.M.; Mader, P.; de Luca, L.; Ferro, S.; Supuran, C.T.; Vullo, D.; Brynda, J.; Řezáčová, P.; Chimirri, A. Synthesis, structure–activity relationship studies, and X-ray crystallographic analysis of arylsulfonamides as potent carbonic anhydrase inhibitors. J. Med. Chem 2012, 55, 3891–3899. [Google Scholar]
- Good, A.C.; Liu, J.; Hirth, B.; Asmussen, G.; Xiang, Y.; Biemann, H.-P.; Bishop, K.A.; Fremgen, T.; Fitzgerald, M.; Gladysheva, T.; et al. Implications of promiscuous Pim-1 kinase fragment inhibitor hydrophobic interactions for fragment-based drug design. J. Med. Chem 2012, 55, 2641–2648. [Google Scholar]
- Patel, S.; Vuillard, L.; Cleasby, A.; Murray, C.W.; Yon, J. Apo and inhibitor complex structures of BACE (β-secretase). J. Mol. Biol 2004, 343, 407–416. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [Google Scholar]
- Torres, F.E.; Kuhn, P.; de Bruyker, D.; Bell, A.G.; Wolkin, M.V.; Peeters, E.; Williamson, J.R.; Anderson, G.B.; Schmitz, G.P.; Recht, M.I.; et al. Enthalpy arrays. Proc. Natl. Acad. Sci. USA 2004, 101, 9517–9522. [Google Scholar]
- Drinkwater, N.; Vu, H.; Lovell, K.M.; Criscione, K.R.; Collins, B.M.; Prisinzano, T.E.; Poulsen, S.A.; McLeish, M.J.; Grunewald, G.L.; Martin, J.L. Fragment-based screening by X-ray crystallography, MS and isothermal titration calorimetry to identify PNMT (phenylethanolamine N-methyltransferase) inhibitors. Biochem. J 2010, 431, 51–61. [Google Scholar]
- Pearl, L.H.; Prodromou, C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem 2006, 75, 271–294. [Google Scholar]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar]
- Cheung, C.H.; Chen, H.H.; Cheng, L.T.; Lyu, K.W.; Kanwar, J.R.; Chang, J.Y. Targeting Hsp90 with small molecule inhibitors induces the over-expression of the anti-apoptotic molecule, survivin, in human A549, HONE-1 and HT-29 cancer cells. Mol. Cancer 2010, 9, 77. [Google Scholar]
- Lu, X.; Xiao, L.; Wang, L.; Ruden, D.M. Hsp90 inhibitors and drug resistance in cancer: The potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol 2012, 83, 995–1004. [Google Scholar]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res 2012, 18, 64–76. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Article title | Target protein | Primary/secondary FBDD screening method | Binding/activity assay |
|---|---|---|---|---|
| [54,55] | Application of Fragment Screening by X-ray Crystallography to the Discovery of Aminopyridines as Inhibitors of -Secretase | β-Secretase | X-ray crystallography | Fluorescence-based activity assay |
| [56] | Missing fragments: detecting cooperative binding in fragment-based drug design | hPNMT | X-ray crystallography | ITC/Molecular dynamics free energy calculation |
| [57] | Fragment-based screening for inhibitors of PDE4A using enthalpy arrays and X-ray crystallography | Phosphodiesterase 4A | High-throughput calorimetry/X-ray crystallography | High-throughput calorimetry |
| [58,59] | Fragment-Based Drug Discovery Applied to Hsp90. Discovery of Two Lead Series with High Ligand Efficiency | Hsp90 | NMR/X-ray crystallography | ITC/Bioassay |
| [60] | Fragment-Based Discovery of Bromodomain Inhibitors Part 1: Inhibitor binding modes and implications for lead discovery | Bromodomain | Fluorescence anisotropy assay/X-ray crystallography | Fluorescence anisotropy assay |
| [61] | Fragment-Based Discovery of Bromodomain Inhibitors Part 2: Optimization of Phenylisoxazole Sulfonamide | Bromodomain/AcK pocket | Fluorescence anisotropy assay/Modelling X-ray crystallography | SPR/Thermal shift assay |
| [62] | Structure-based design of potent and ligand-efficient inhibitors of CTX-M class A β-lactamase | β-lactamase CTX-M | Docking/X-ray crystallography | UV-absorbance based bioassays/Antibacterial activity |
| [63] | Discovery of 1,2,4-triaine derivatives as adenosine A2A antagonists using structure based drug design | Adenosine A2 receptor | Docking/X-ray crystallography | SPR |
| [64] | Discovery and Optimization of New Benzimidazole- and Benzoxazole-Pyrimidone Selective PI3Kβ Inhibitors for the Treatment of Phosphatase and TENsin homologue (PTEN)-Deficient Cancers | PI3K | In vitro enzyme assay/Cell based assay X-ray crystallography | In vitro enzyme assay/Cell-based assay |
| [65] | Synthesis, Structure–Activity Relationship Studies, and X-ray Crystallographic Analysis of Arylsulfonamides as Potent Carbonic Anhydrase Inhibitor | Carbonic anhydrases | Docking/X-ray crystallography | Stopped-flow kinetic assay |
| [66] | Implications of Promiscuous Pim-1 Kinase Fragment Inhibitor Hydrophobic Interactions for Fragment-Based Drug Design | Pim-1 Kinase | Docking/X-ray crystallography | Mobility shift assay |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chilingaryan, Z.; Yin, Z.; Oakley, A.J. Fragment-Based Screening by Protein Crystallography: Successes and Pitfalls. Int. J. Mol. Sci. 2012, 13, 12857-12879. https://doi.org/10.3390/ijms131012857
Chilingaryan Z, Yin Z, Oakley AJ. Fragment-Based Screening by Protein Crystallography: Successes and Pitfalls. International Journal of Molecular Sciences. 2012; 13(10):12857-12879. https://doi.org/10.3390/ijms131012857
Chicago/Turabian StyleChilingaryan, Zorik, Zhou Yin, and Aaron J. Oakley. 2012. "Fragment-Based Screening by Protein Crystallography: Successes and Pitfalls" International Journal of Molecular Sciences 13, no. 10: 12857-12879. https://doi.org/10.3390/ijms131012857