Probing Nucleic Acid Interactions and Pre-mRNA Splicing by Förster Resonance Energy Transfer (FRET) Microscopy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
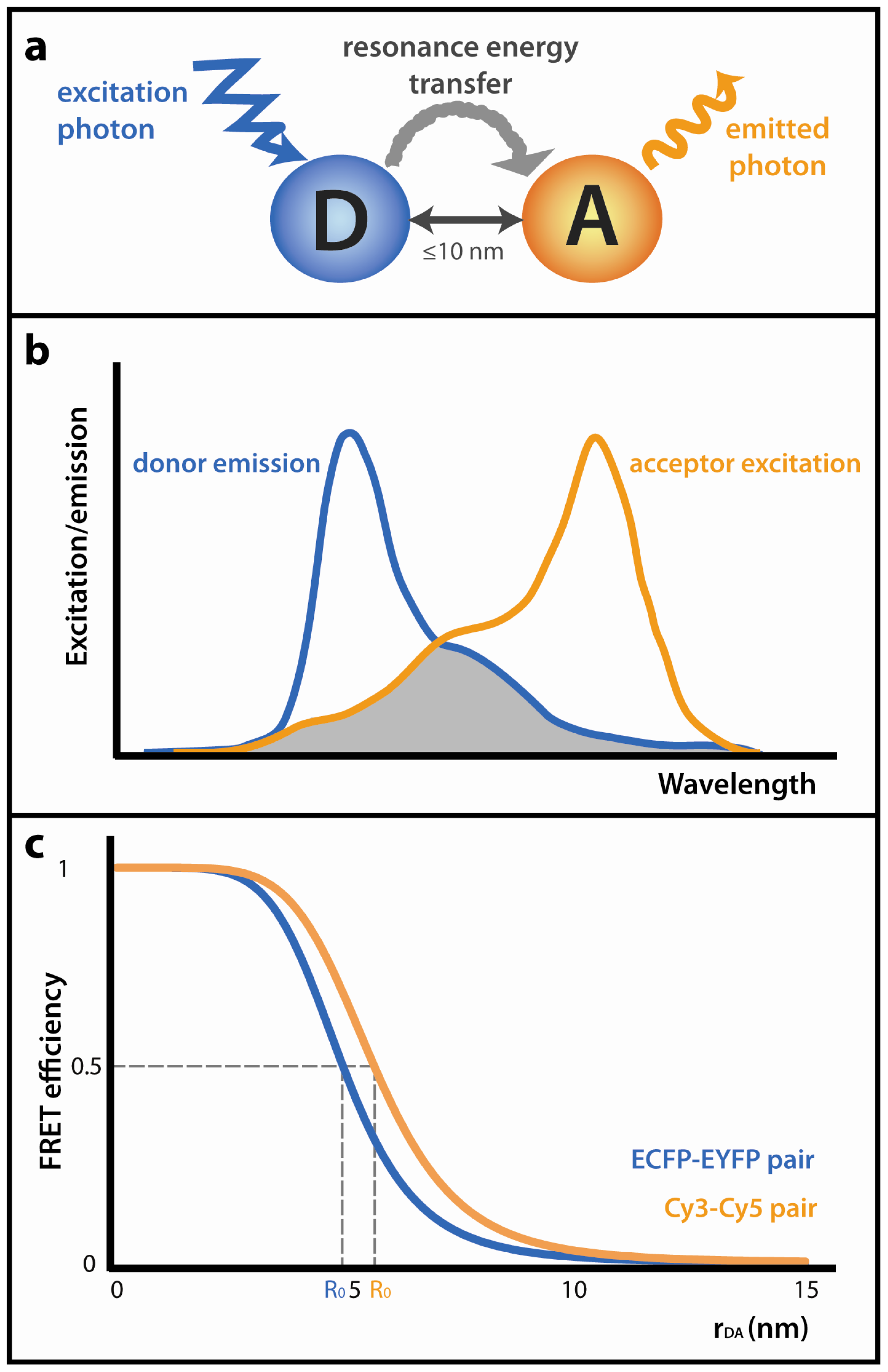
:1. Introduction
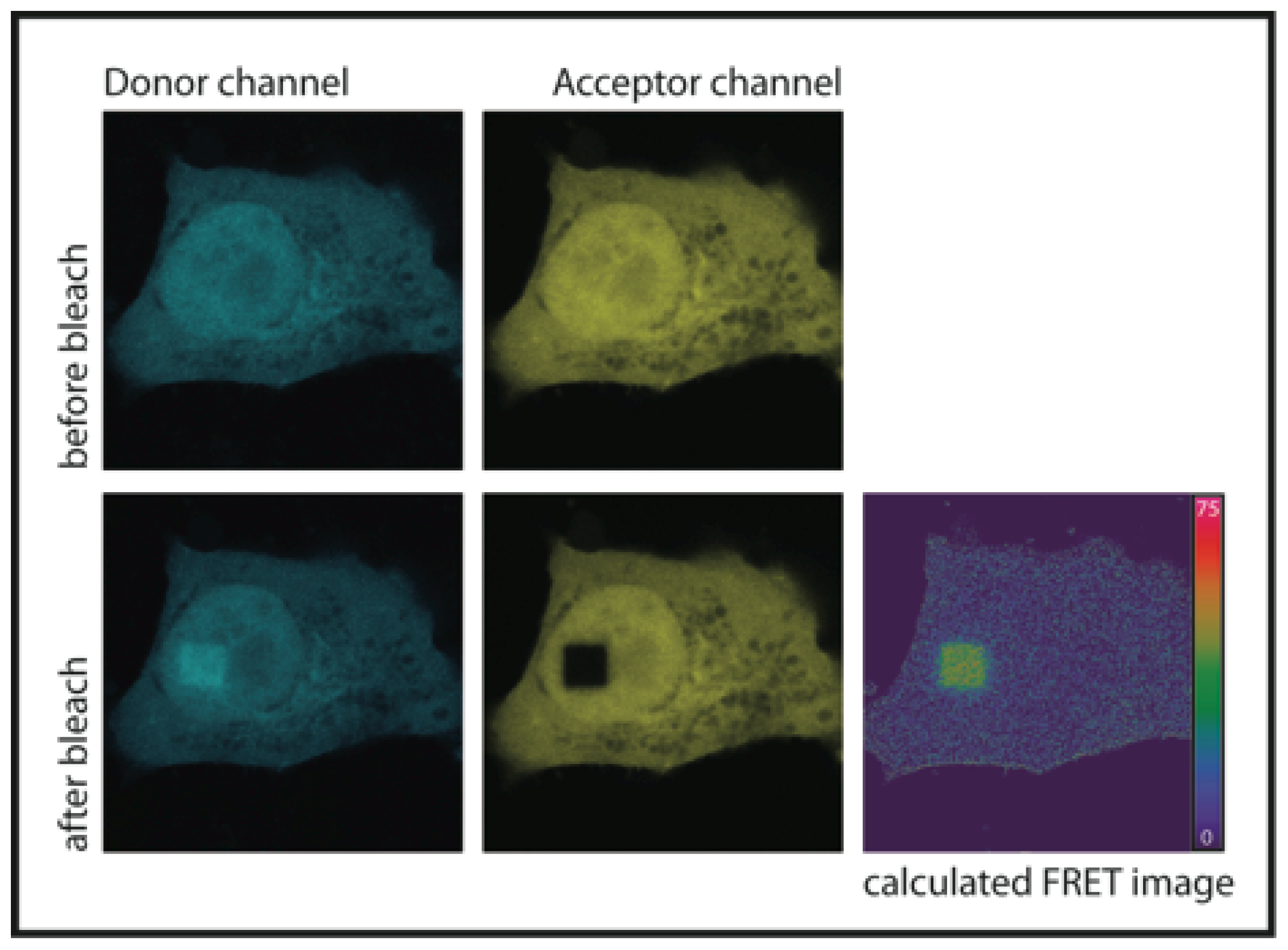
1.1. Acceptor Photobleaching FRET (AB-FRET)
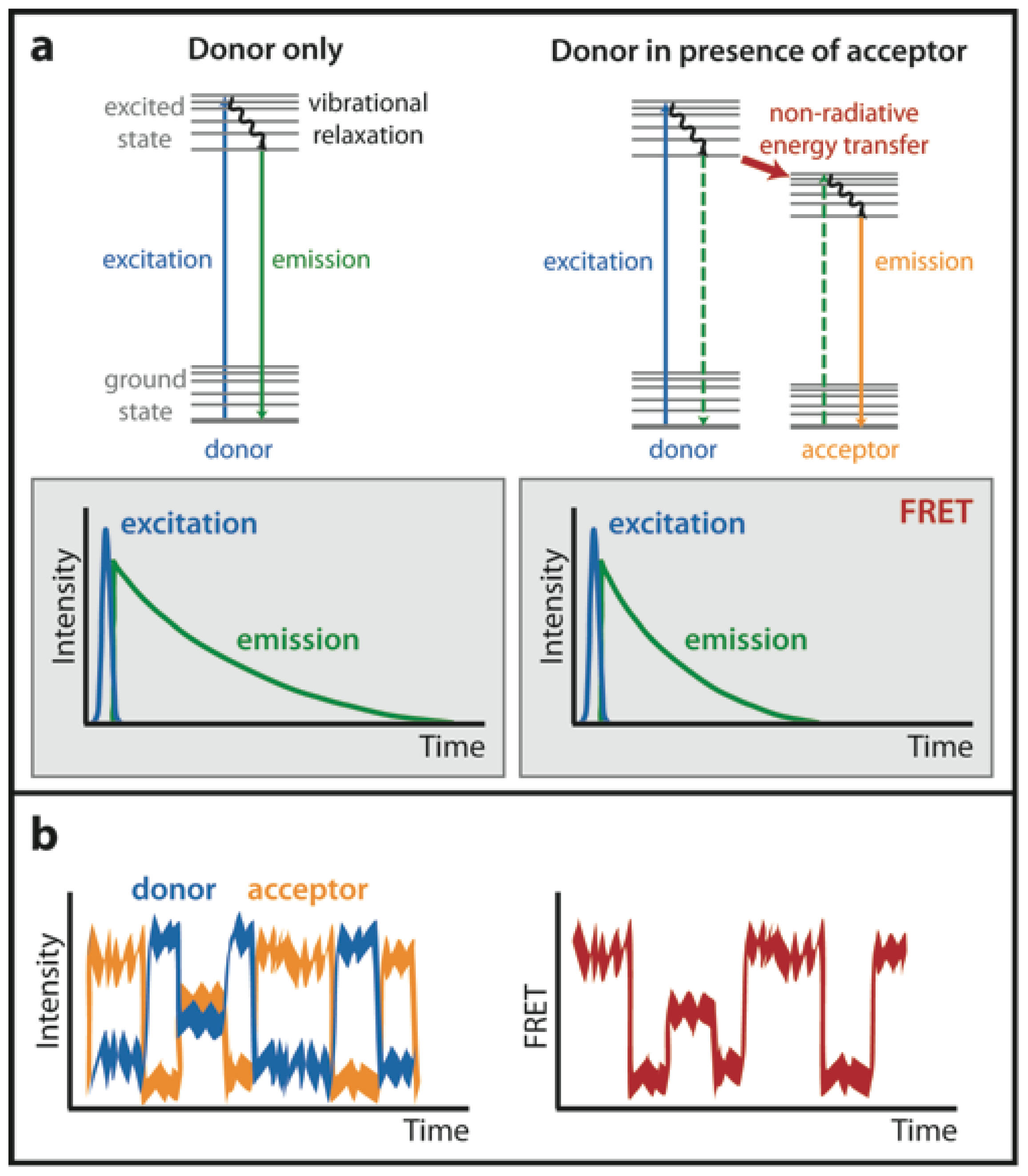
1.2. Fluorescence Lifetime Imaging Microscopy (FLIM)
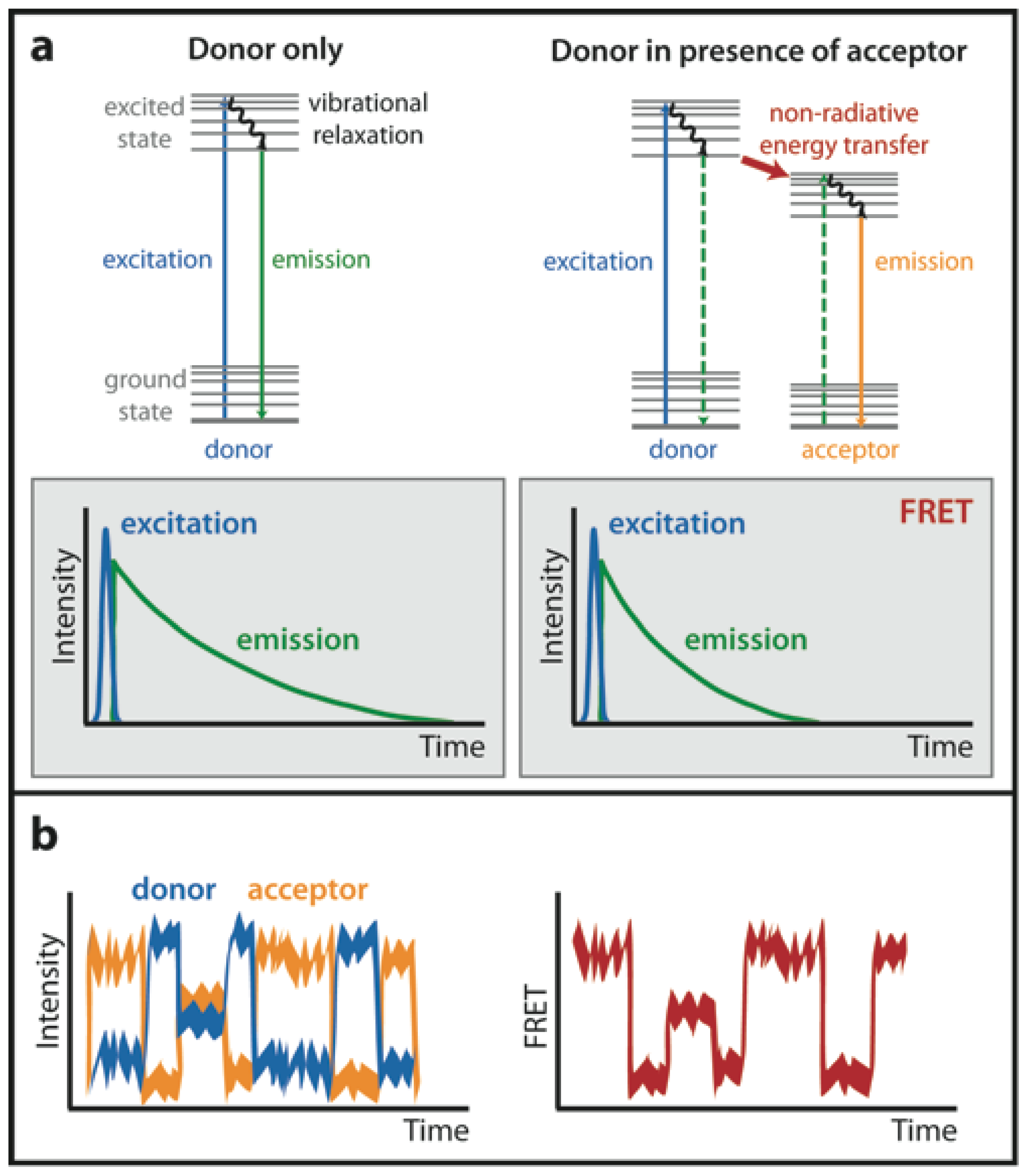
1.4. Single-Molecule FRET (smFRET)
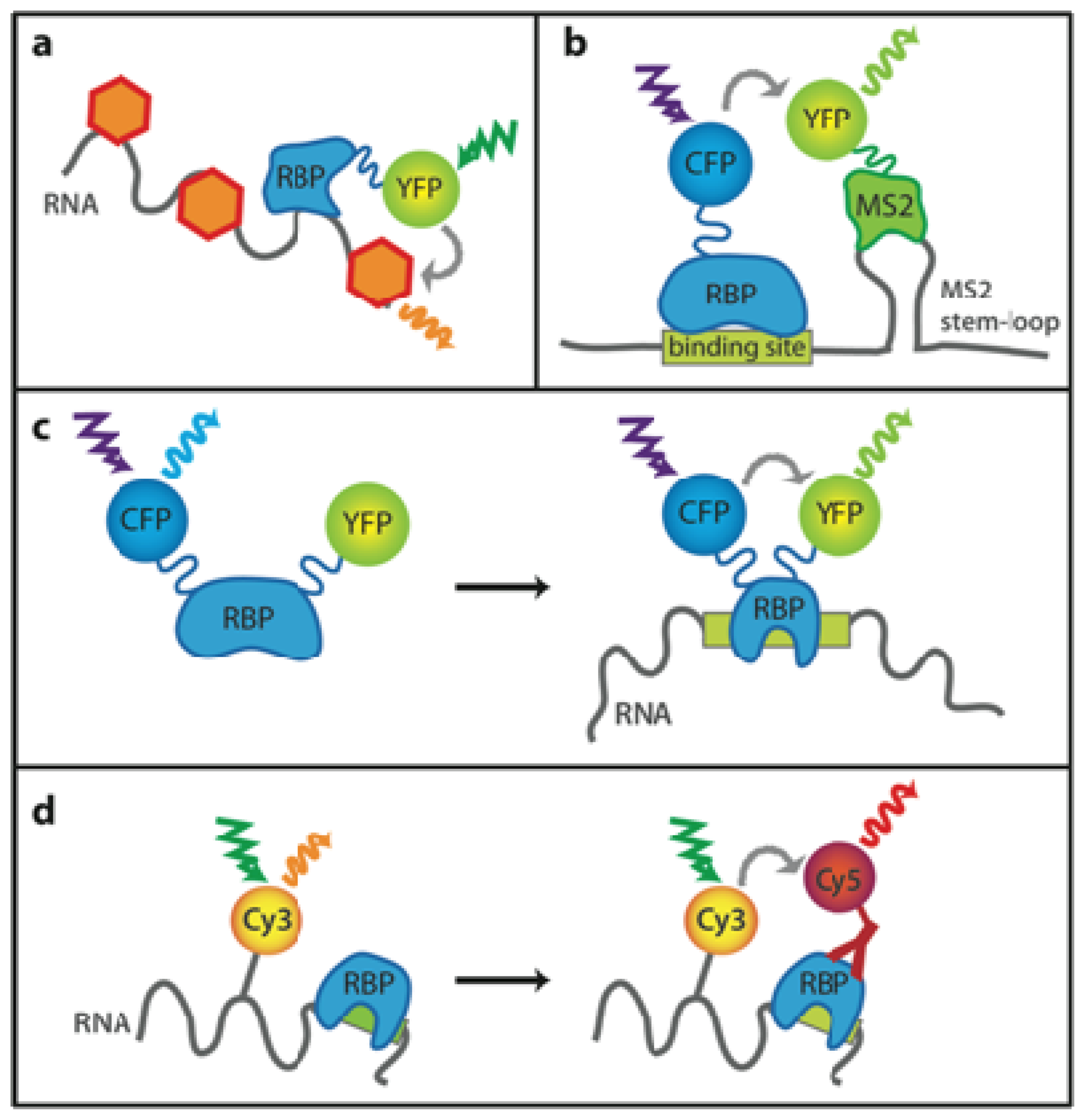
2. Detection of Nucleic Acid-Protein Interactions in situ
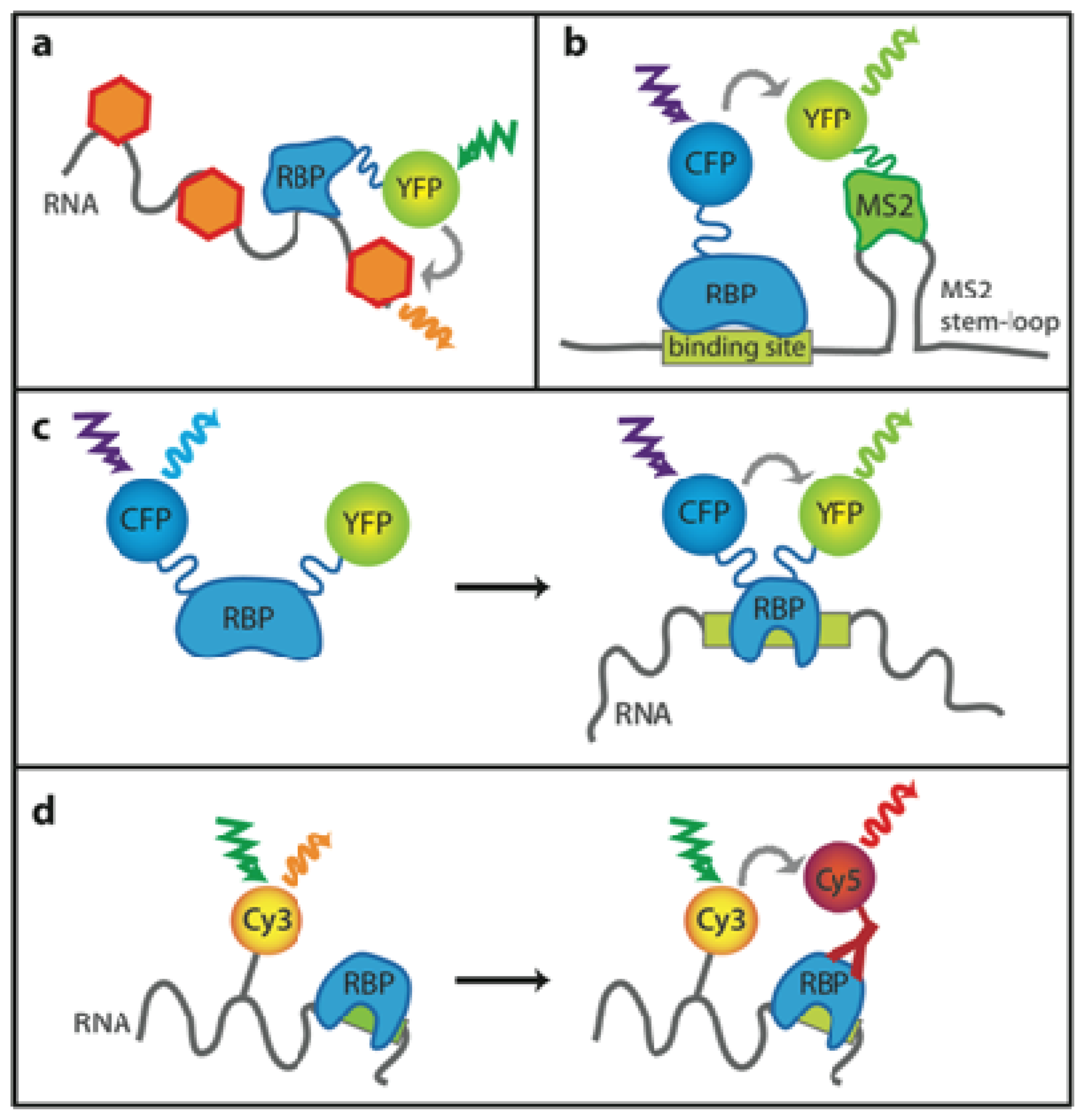
2.1. Unspecific Labeling of Nucleic Acids
2.2. Monitoring Sequence-Specific Binding of Proteins
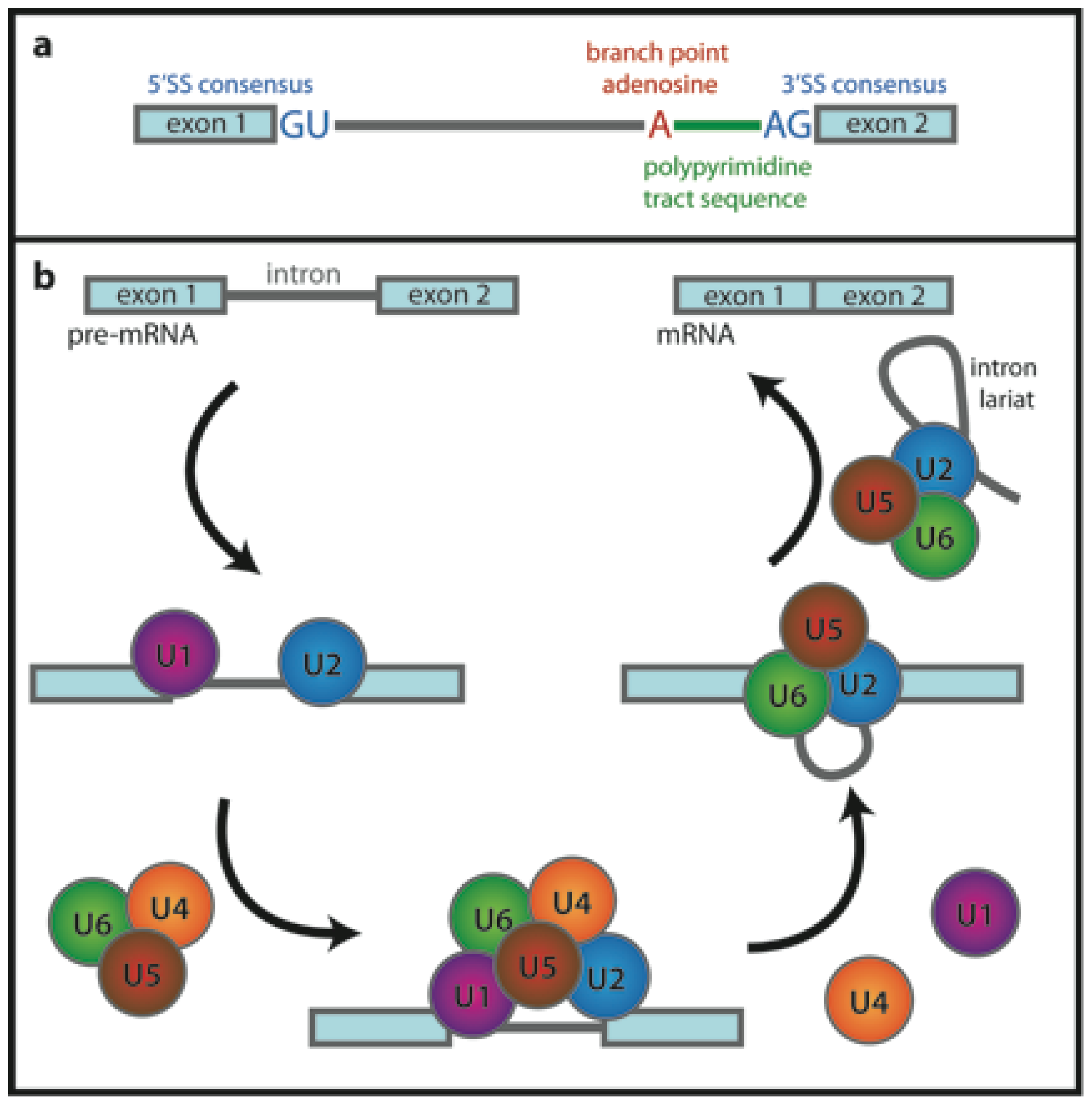
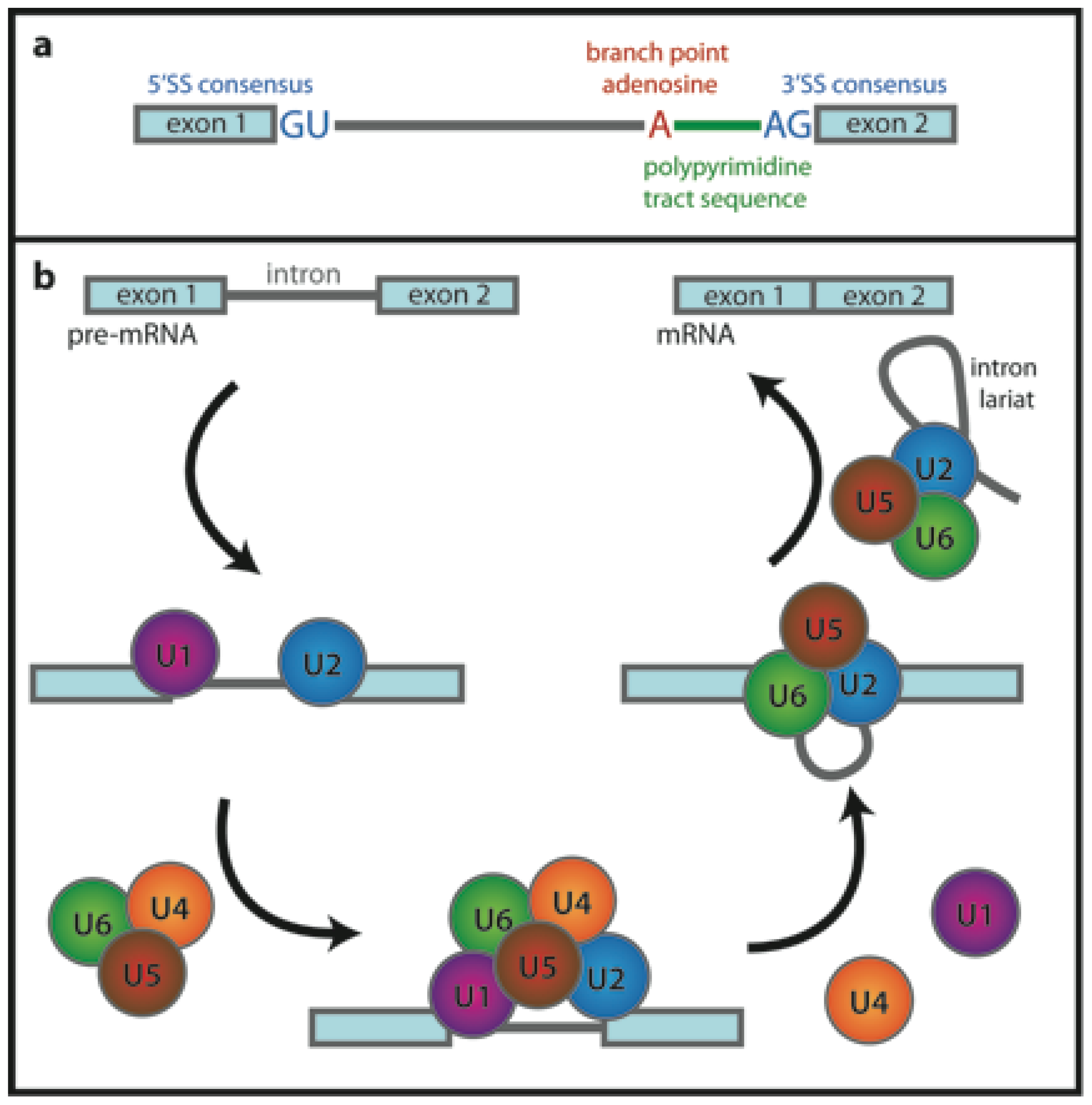
3. Spliceosome, A Case Study of RNA Interactions Investigated by FRET
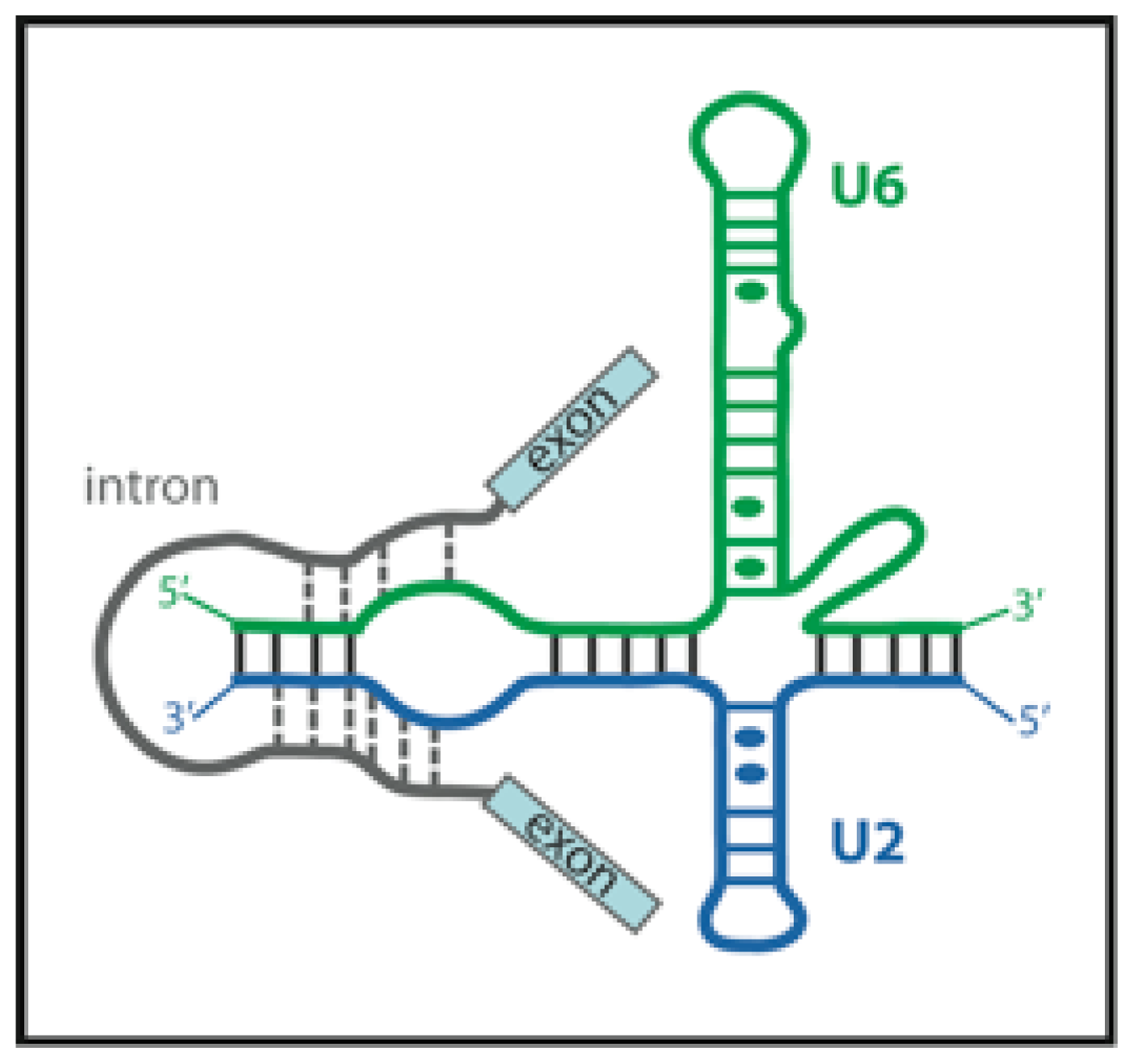
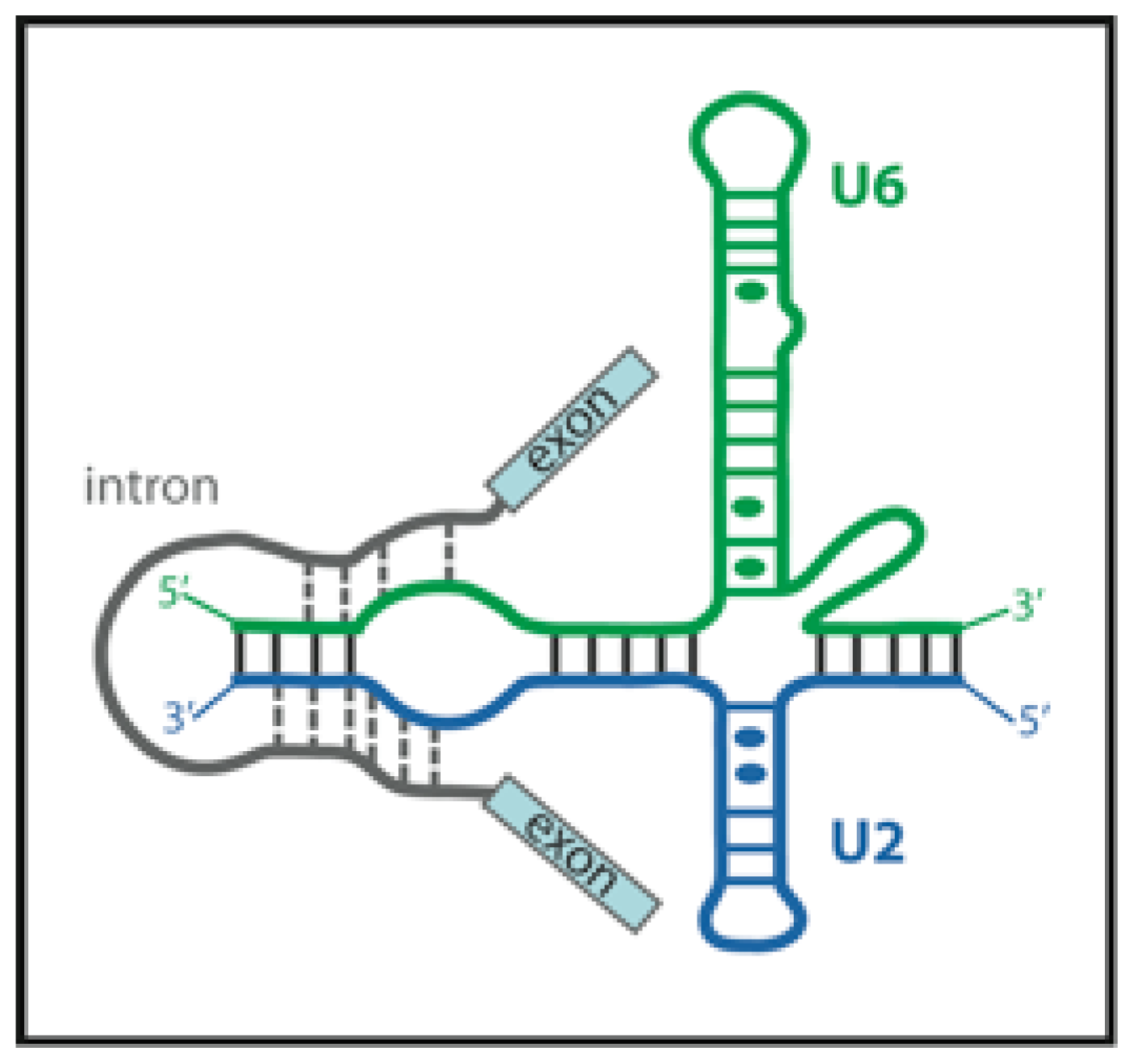
3.1. RNA Conformational Dynamics
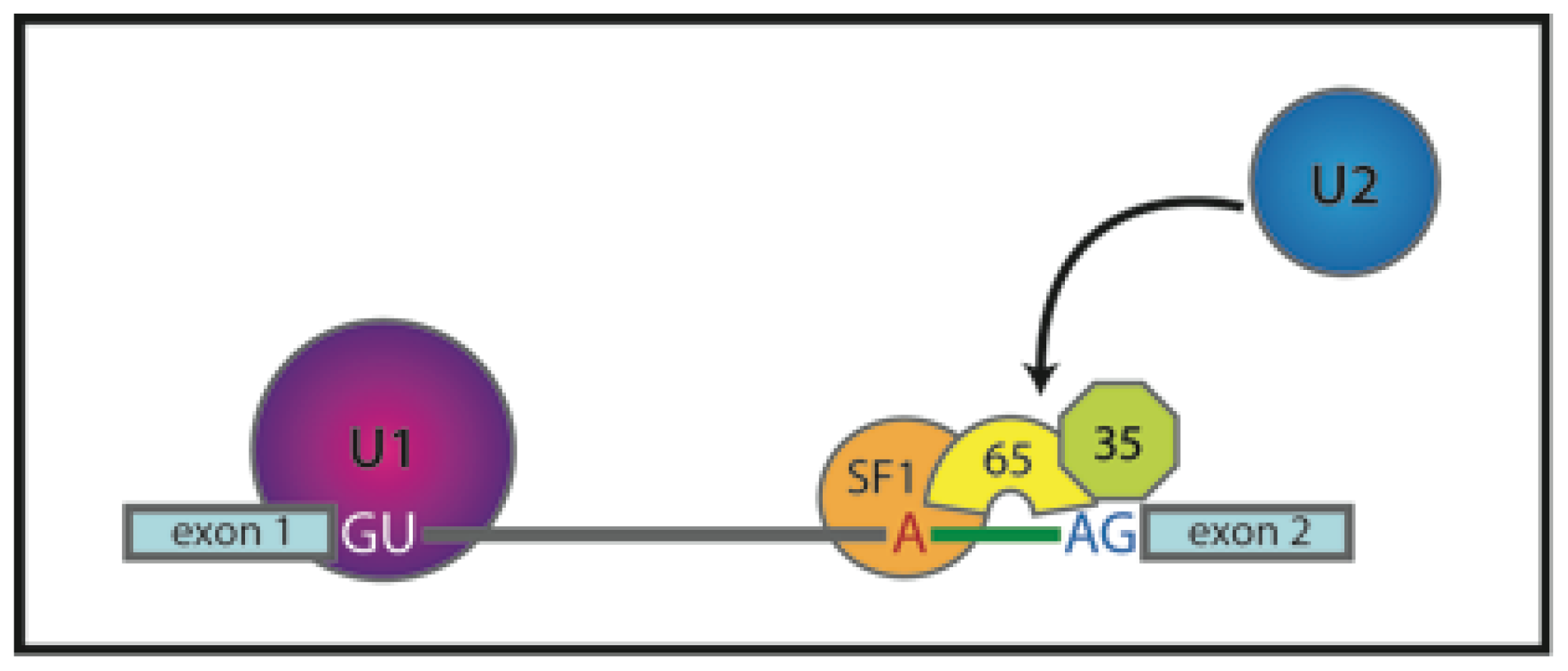
3.2. Early Complex Assembly
3.3. SR-Protein Interactions during Intron Recognition
3.4. Formation of the U4/U6·U5 Tri-snRNP
3.5. FRET Helps to Solve the Catalytic core Structure and Dynamics
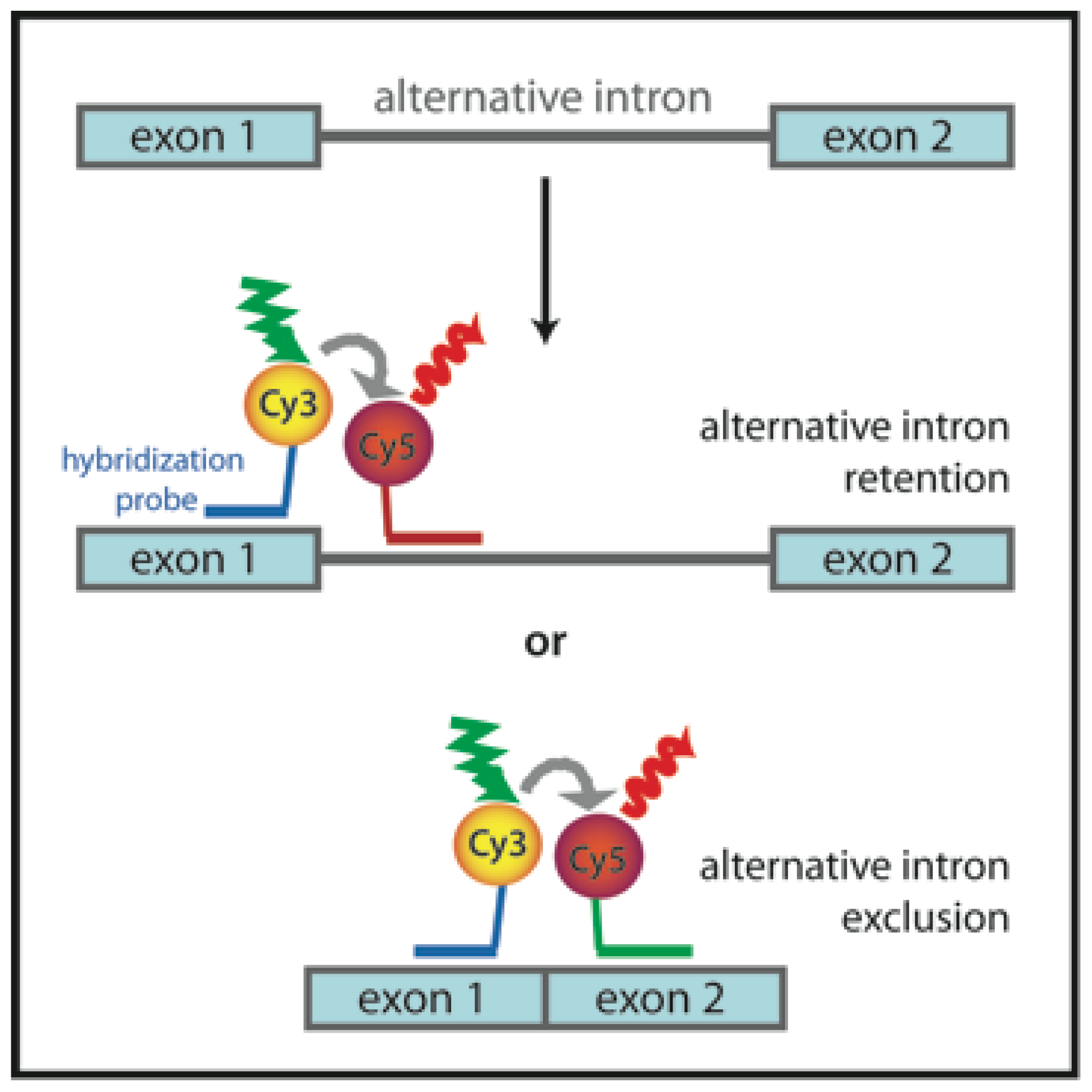
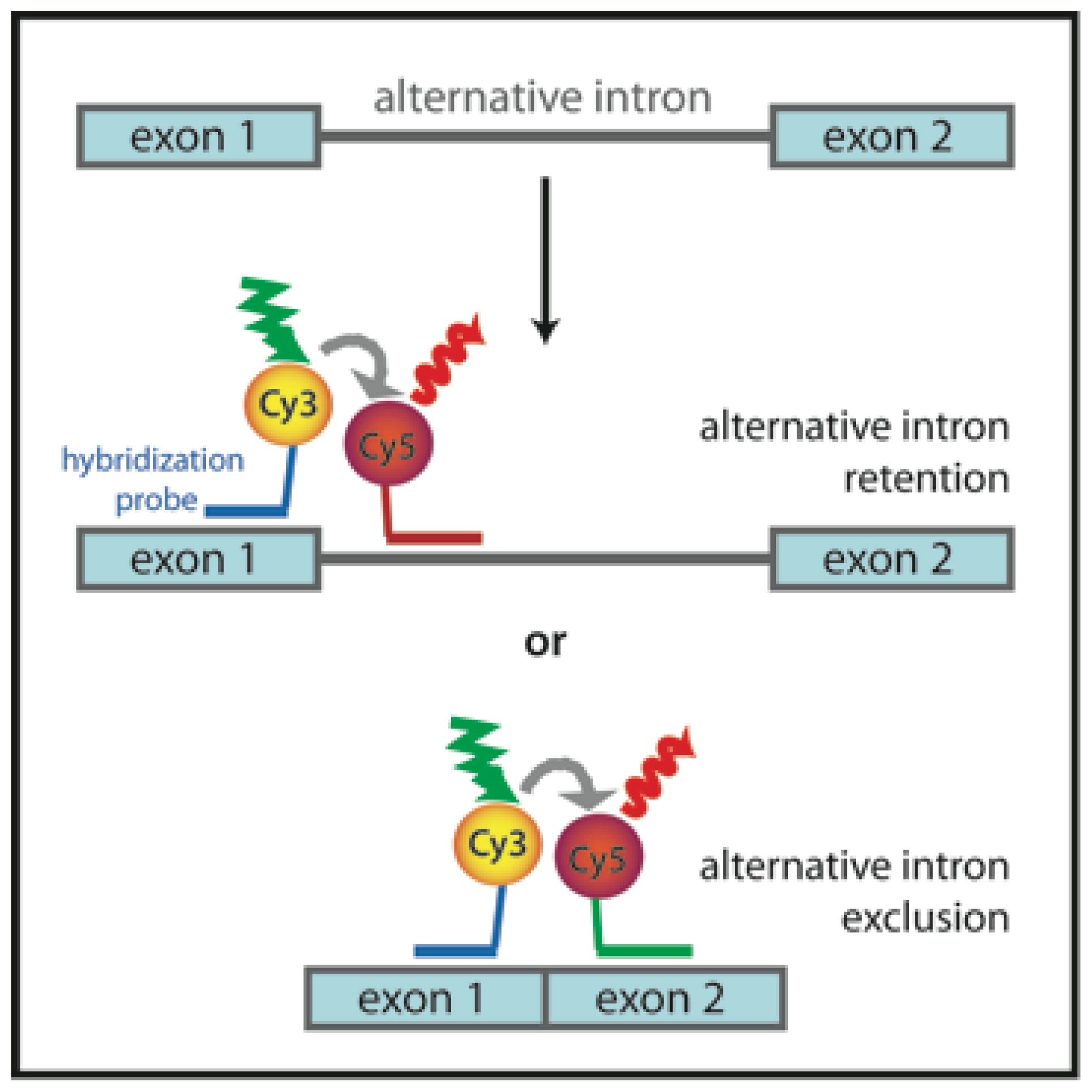
3.6. Probing Alternative Splicing with FRET
4. Conclusion and Outlook
Acknowledgments
References
- Förster, T. Energiewanderung und Fluoreszenz. Naturwissenschaften 1946, 33, 166–175. [Google Scholar]
- Sapsford, K.E.; Berti, L.; Medintz, I.L. Materials for fluorescence resonance energy transfer analysis: Beyond traditional donor-acceptor combinations. Angew. Chem. Int. Ed. Engl 2006, 45, 4562–4589. [Google Scholar]
- Gonçalves, M.S.T. Fluorescent labeling of biomolecules with organic probes. Chem. Rev 2009, 109, 190–212. [Google Scholar]
- Stepanenko, O.V.; Shcherbakova, D.M.; Kuznetsova, I.M.; Turoverov, K.K.; Verkhusha, V.V. Modern fluorescent proteins: From chromophore formation to novel intracellular applications. Biotechniques 2011, 51, 313–327. [Google Scholar]
- Jares-Erijman, E.A.; Jovin, T.M. FRET imaging. Nat. Biotechnol 2003, 21, 1387–1395. [Google Scholar]
- Galbraith, C.G.; Galbraith, J.A. Super-resolution microscopy at a glance. J. Cell Sci 2011, 124, 1607–1611. [Google Scholar]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol 2010, 190, 165–175. [Google Scholar]
- Grecco, H.E.; Verveer, P.J. FRET in cell biology: Still shining in the age of super-resolution? Chemphyschem 2011, 12, 484–490. [Google Scholar]
- Piston, D.W.; Kremers, G.-J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci 2007, 32, 407–414. [Google Scholar]
- Pietraszewska-Bogiel, A.; Gadella, T.W.J. FRET microscopy: From principle to routine technology in cell biology. J. Microsc 2011, 241, 111–118. [Google Scholar]
- Patterson, G.H.; Piston, D.W.; Barisas, B.G. Förster distances between green fluorescent protein pairs. Anal. Biochem 2000, 284, 438–440. [Google Scholar]
- Berney, C.; Danuser, G. FRET or no FRET: A quantitative comparison. Biophys. J 2003, 84, 3992–4010. [Google Scholar]
- Malkani, N.; Schmid, J.A. Some secrets of fluorescent proteins: distinct bleaching in various mounting fluids and photoactivation of cyan fluorescent proteins at YFP-excitation. PLoS One 2011, 6, e18586. [Google Scholar]
- Nienhaus, G.U.; Wiedenmann, J. Structure, dynamics and optical properties of fluorescent proteins: Perspectives for marker development. Chemphyschem 2009, 10, 1369–1379. [Google Scholar]
- Dickson, R.M.; Cubitt, A.B.; Tsien, R.Y.; Moerner, W.E. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 1997, 388, 355–358. [Google Scholar]
- Valentin, G.; Verheggen, C.; Piolot, T.; Neel, H.; Coppey-Moisan, M.; Bertrand, E. Photoconversion of YFP into a CFP-like species during acceptor photobleaching. Nat. Meth 2005, 2, 801. [Google Scholar]
- Thaler, C.; Vogel, S.S.; Ikeda, S.R.; Chen, H. Photobleaching of YFP does not produce a CFP-like species that affects FRET measurements. Nat. Meth 2006, 3, 491. [Google Scholar]
- Verrier, S.E.; Söling, H.-D. Photobleaching of YFP does not produce a CFP-like species that affects FRET measurements. Nat. Meth 2006, 3, 491–492. [Google Scholar]
- Valentin, G.; Verheggen, C.; Piolot, T.; Neel, H.; Zimmermann, T.; Coppey-Moisan, M.; Bertrand, E. Photobleaching of YFP does not produce a CFP-like species that affects FRET measurements. Nat. Meth 2006, 3, 492–493. [Google Scholar]
- Kirber, M.T.; Chen, K.; Keaney, J.F. YFP photoconversion revisited: Confirmation of the CFP-like species. Nat. Meth 2007, 4, 767–768. [Google Scholar]
- Becker, W. Fluorescence lifetime imaging—Techniques and applications. J. Microsc 2012, 247, 119–136. [Google Scholar]
- Becker, W.; Bergmann, A.; Hink, M.A.; König, K.; Benndorf, K.; Biskup, C. Fluorescence lifetime imaging by time-correlated single-photon counting. Micro. Res. Tech 2004, 63, 58–66. [Google Scholar]
- Levitt, J.A.; Matthews, D.R.; Ameer-Beg, S.M.; Suhling, K. Fluorescence lifetime and polarization-resolved imaging in cell biology. Curr. Opin. Biotechnol 2009, 20, 28–36. [Google Scholar]
- Van Munster, E.B.; Gadella, T.W.J. Fluorescence Lifetime Imaging Microscopy (FLIM). Adv. Biochem. Eng. Biotechnol 2005, 95, 143–175. [Google Scholar]
- Sun, Y.; Day, R.N.; Periasamy, A. Investigating protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Nat. Protoc 2011, 6, 1324–1340. [Google Scholar]
- Ha, T.; Enderle, T.; Ogletree, D.F.; Chemla, D.S.; Selvin, P.R.; Weiss, S. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA 1996, 93, 6264–6268. [Google Scholar]
- Tinoco, I.; Gonzalez, R.L. Biological mechanisms, one molecule at a time. Genes Dev 2011, 25, 1205–1231. [Google Scholar]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Meth 2008, 5, 507–516. [Google Scholar]
- Tinoco, I.; Chen, G.; Qu, X. RNA reactions one molecule at a time. Cold Spring Harb. Perspect. Biol 2010, 2, a003624. [Google Scholar]
- Sakon, J.J.; Weninger, K.R. Detecting the conformation of individual proteins in live cells. Nat. Meth 2010, 7, 203–205. [Google Scholar]
- Cremazy, F.G.E.; Manders, E.M.M.; Bastiaens, P.I.H.; Kramer, G.; Hager, G.L.; van Munster, E.B.; Verschure, P.J.; Gadella, T.J.; van Driel, R. Imaging in situ protein-DNA interactions in the cell nucleus using FRET-FLIM. Exp. Cell. Res 2005, 309, 390–396. [Google Scholar]
- Lorenz, M. Visualizing protein—RNA interactions inside cells by fluorescence resonance energy transfer. RNA 2009, 15, 97–103. [Google Scholar]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol 2002, 20, 87–90. [Google Scholar]
- Sapra, A.K.; Ankö, M.-L.; Grishina, I.; Lorenz, M.; Pabis, M.; Poser, I.; Rollins, J.; Weiland, E.-M.; Neugebauer, K.M. SR protein family members display diverse activities in the formation of nascent and mature mRNPs in vivo. Mol. Cell 2009, 34, 179–190. [Google Scholar]
- Meades, G.; Benson, B.K.; Grove, A.; Waldrop, G.L. A tale of two functions: Enzymatic activity and translational repression by carboxyltransferase. Nucleic Acids Res 2010, 38, 1217–1227. [Google Scholar]
- Huranová, M.; Jablonski, J.A.; Benda, A.; Hof, M.; Stanek, D.; Caputi, M. In vivo detection of RNA-binding protein interactions with cognate RNA sequences by fluorescence resonance energy transfer. RNA 2009, 15, 2063–2071. [Google Scholar]
- Endoh, T.; Funabashi, H.; Mie, M.; Kobatake, E. Method for detection of specific nucleic acids by recombinant protein with fluorescent resonance energy transfer. Anal. Chem 2005, 77, 4308–4314. [Google Scholar]
- Endoh, T.; Mie, M.; Kobatake, E. Direct detection of RNA transcription by FRET imaging using fluorescent protein probe. J. Biotechnol 2008, 133, 413–417. [Google Scholar]
- Gerecht, P.S.D.; Taylor, M.A.; Port, J.D. Intracellular localization and interaction of mRNA binding proteins as detected by FRET. BMC Cell Biol 2010, 11, 69. [Google Scholar]
- Brody, E.; Abelson, J. The “spliceosome”: Yeast pre-messenger RNA associates with a 40S complex in a splicing-dependent reaction. Science 1985, 228, 963–967. [Google Scholar]
- Bindereif, A.; Green, M.R. An ordered pathway of snRNP binding during mammalian pre-mRNA splicing complex assembly. EMBO J 1987, 6, 2415–2424. [Google Scholar]
- Rino, J.; Carmo-Fonseca, M. The spliceosome: A self-organized macromolecular machine in the nucleus? Trends Cell Biol 2009, 19, 375–384. [Google Scholar]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol 2011, 3, a003707. [Google Scholar]
- Abelson, J.; Blanco, M.; Ditzler, M.A.; Fuller, F.; Aravamudhan, P.; Wood, M.; Villa, T.; Ryan, D.E.; Pleiss, J.A.; Maeder, C.; et al. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat. Struct. Mol. Biol 2010, 17, 504–512. [Google Scholar]
- Zamore, P.D.; Patton, J.G.; Green, M.R. Cloning and domain structure of the mammalian splicing factor U2AF. Nature 1992, 355, 609–614. [Google Scholar]
- Merendino, L.; Guth, S.; Bilbao, D.; Martínez, C.; Valcárcel, J. Inhibition of msl-2 splicing by Sex-lethal reveals interaction between U2AF35 and the 3′ splice site AG. Nature 1999, 402, 838–841. [Google Scholar]
- Chusainow, J.; Ajuh, P.M.; Trinkle-Mulcahy, L.; Sleeman, J.E.; Ellenberg, J.; Lamond, A.I. FRET analyses of the U2AF complex localize the U2AF35/U2AF65 interaction in vivo and reveal a novel self-interaction of U2AF35. RNA 2005, 11, 1201–1214. [Google Scholar]
- Rino, J.; Desterro, J.M.P.; Pacheco, T.R.; Gadella, T.W.J.; Carmo-Fonseca, M. Splicing factors SF1 and U2AF associate in extraspliceosomal complexes. Mol. Cell. Biol 2008, 28, 3045–3057. [Google Scholar]
- Matlin, A.J.; Clark, F.; Smith, C.W.J. Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol 2005, 6, 386–398. [Google Scholar]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J 2009, 417, 15–27. [Google Scholar]
- Ellis, J.D.; Llères, D.; Denegri, M.; Lamond, A.I.; Cáceres, J.F. Spatial mapping of splicing factor complexes involved in exon and intron definition. J. Cell Biol 2008, 181, 921–934. [Google Scholar]
- Girard, C.; Will, C.L.; Peng, J.; Makarov, E.M.; Kastner, B.; Lemm, I.; Urlaub, H.; Hartmuth, K.; Lührmann, R. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat. Commun 2012, 3, 994. [Google Scholar]
- Nottrott, S.; Urlaub, H.; Lührmann, R. Hierarchical, clustered protein interactions with U4/U6 snRNA: A biochemical role for U4/U6 proteins. EMBO J 2002, 21, 5527–5538. [Google Scholar]
- WoŸniak, A.K.; Nottrott, S.; Kühn-Hölsken, E.; Schröder, G.F.; Grubmüller, H.; Lührmann, R.; Seidel, C.A.M.; Oesterhelt, F. Detecting protein-induced folding of the U4 snRNA kink-turn by single-molecule multiparameter FRET measurements. RNA 2005, 11, 1545–1554. [Google Scholar]
- Staněk, D.; Neugebauer, K.M. Detection of snRNP assembly intermediates in Cajal bodies by fluorescence resonance energy transfer. J. Cell Biol 2004, 166, 1015–1025. [Google Scholar]
- Novotný, I.; Blažíková, M.; Staněk, D.; Herman, P.; Malinsky, J. In vivo kinetics of U4/U6·U5 tri-snRNP formation in Cajal bodies. Mol. Biol. Cell 2011, 22, 513–23. [Google Scholar]
- Schaffert, N.; Hossbach, M.; Heintzmann, R.; Achsel, T.; Lührmann, R. RNAi knockdown of hPrp31 leads to an accumulation of U4/U6 di-snRNPs in Cajal bodies. EMBO J 2004, 23, 3000–3009. [Google Scholar]
- Guo, Z.; Karunatilaka, K.S.; Rueda, D. Single-molecule analysis of protein-free U2–U6 snRNAs. Nat. Struct. Mol. Biol 2009, 16, 1154–1159. [Google Scholar]
- Yuan, F.; Griffin, L.; Phelps, L.; Buschmann, V.; Weston, K.; Greenbaum, N.L. Use of a novel Förster resonance energy transfer method to identify locations of site-bound metal ions in the U2–U6 snRNA complex. Nucleic Acids Res 2007, 35, 2833–2845. [Google Scholar]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet 2008, 40, 1413–1415. [Google Scholar]
- Blanco, A.M.; Rausell, L.; Aguado, B.; Perez-Alonso, M.; Artero, R. A FRET-based assay for characterization of alternative splicing events using peptide nucleic acid fluorescence in situ hybridization. Nucleic Acids Res 2009, 37, e116. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Šimková, E.; Staněk, D. Probing Nucleic Acid Interactions and Pre-mRNA Splicing by Förster Resonance Energy Transfer (FRET) Microscopy. Int. J. Mol. Sci. 2012, 13, 14929-14945. https://doi.org/10.3390/ijms131114929
Šimková E, Staněk D. Probing Nucleic Acid Interactions and Pre-mRNA Splicing by Förster Resonance Energy Transfer (FRET) Microscopy. International Journal of Molecular Sciences. 2012; 13(11):14929-14945. https://doi.org/10.3390/ijms131114929
Chicago/Turabian StyleŠimková, Eva, and David Staněk. 2012. "Probing Nucleic Acid Interactions and Pre-mRNA Splicing by Förster Resonance Energy Transfer (FRET) Microscopy" International Journal of Molecular Sciences 13, no. 11: 14929-14945. https://doi.org/10.3390/ijms131114929
APA StyleŠimková, E., & Staněk, D. (2012). Probing Nucleic Acid Interactions and Pre-mRNA Splicing by Förster Resonance Energy Transfer (FRET) Microscopy. International Journal of Molecular Sciences, 13(11), 14929-14945. https://doi.org/10.3390/ijms131114929