Eclipsed Acetaldehyde as a Precursor for Producing Vinyl Alcohol

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
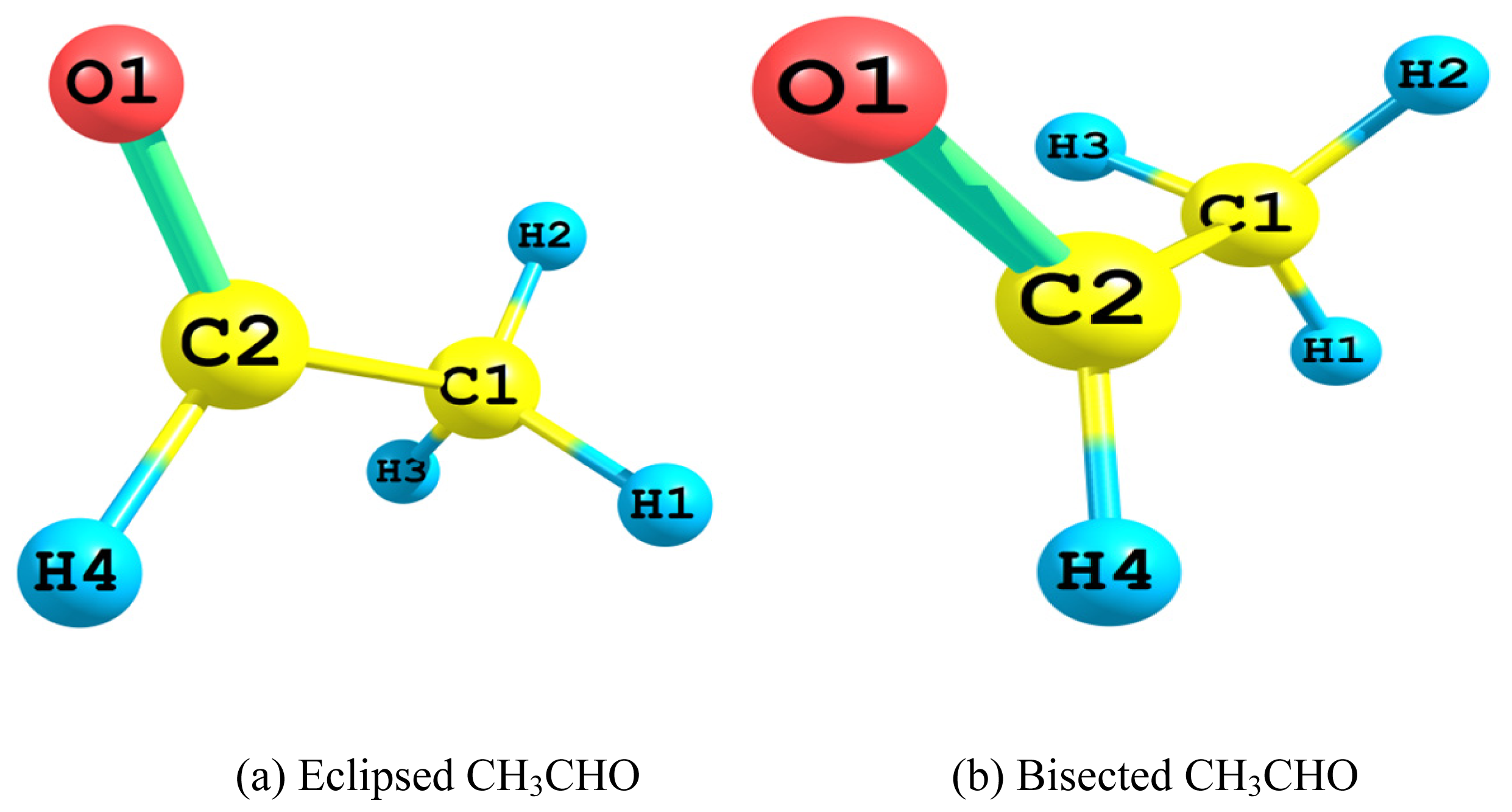
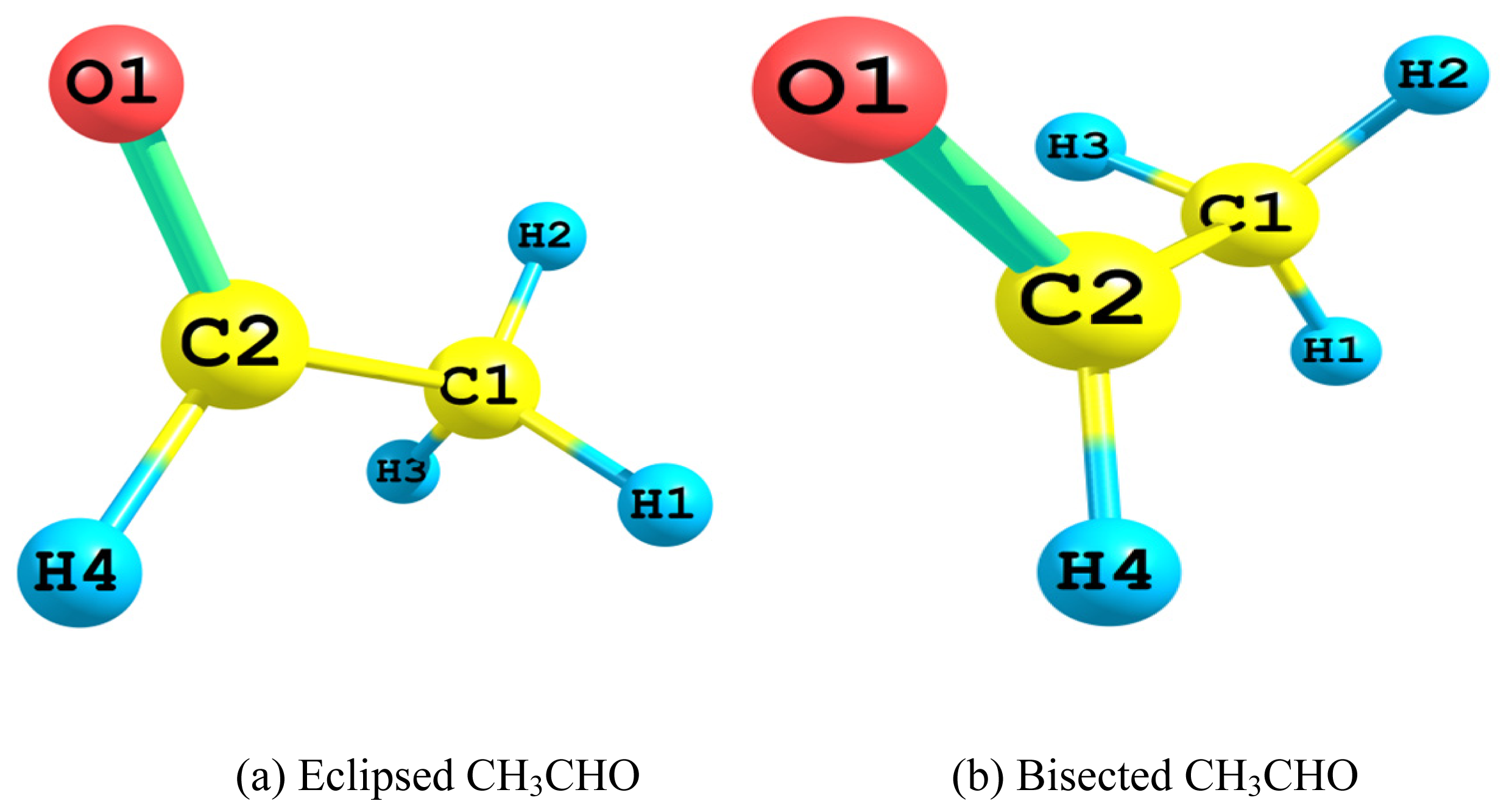
2.1. Relative Stability of Eclipsed and Bisected Acetaldehydes
2.1.1. Relative Energy
2.1.2. Hyperconjugative Interactions
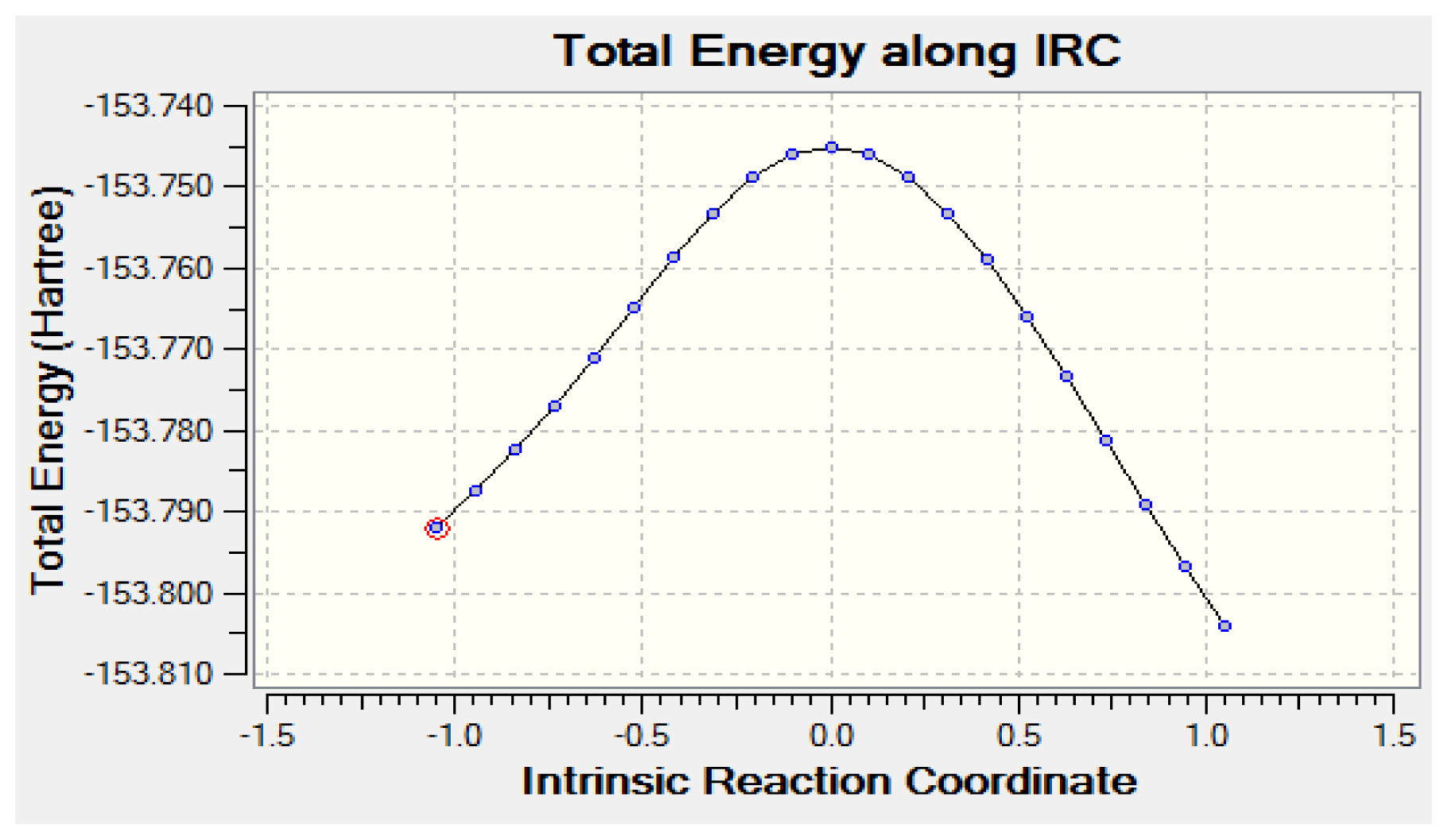
2.2. Tautomerization Reaction of Eclipsed Acetaldehyde and Vinyl Alcohol
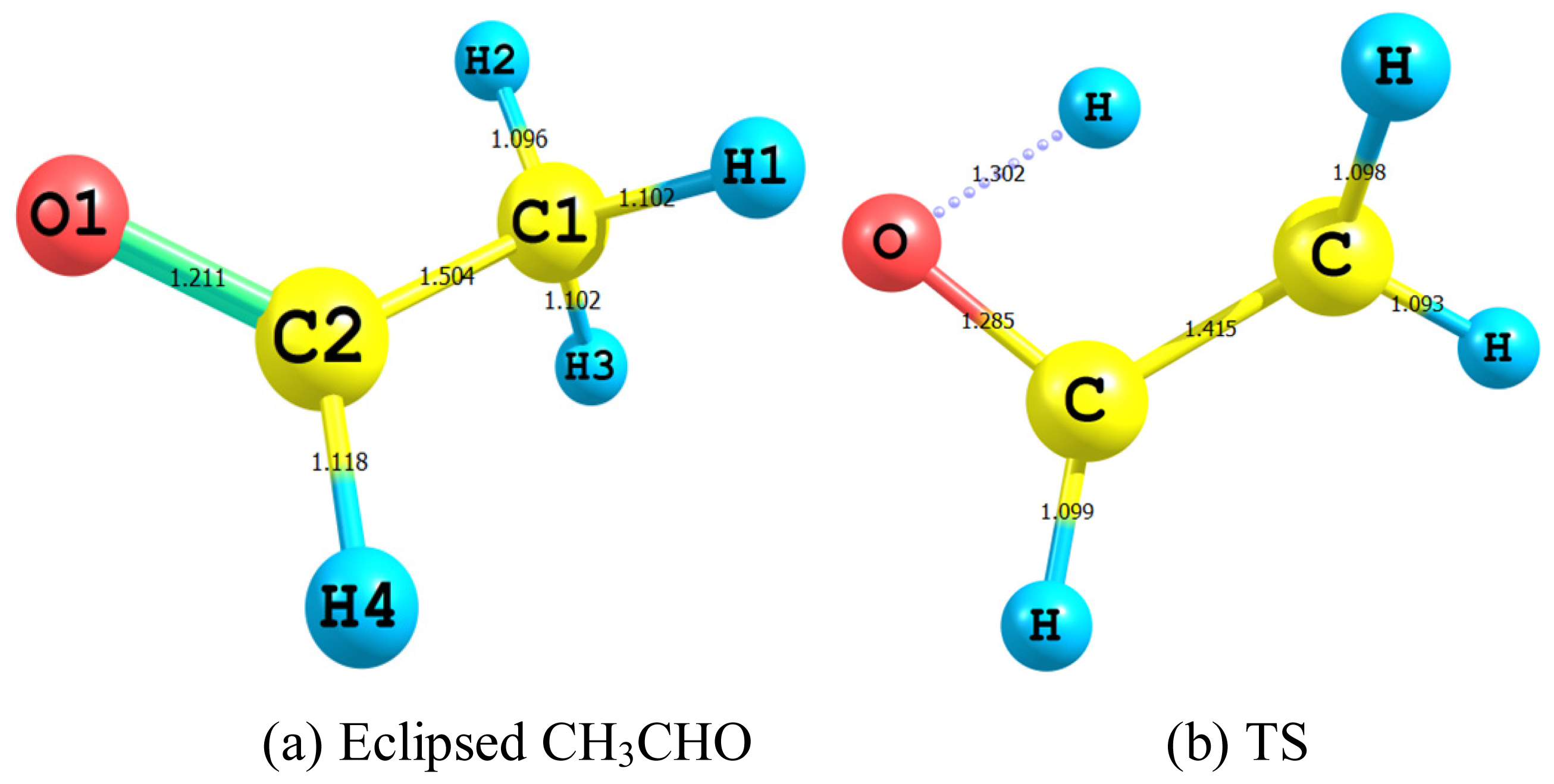
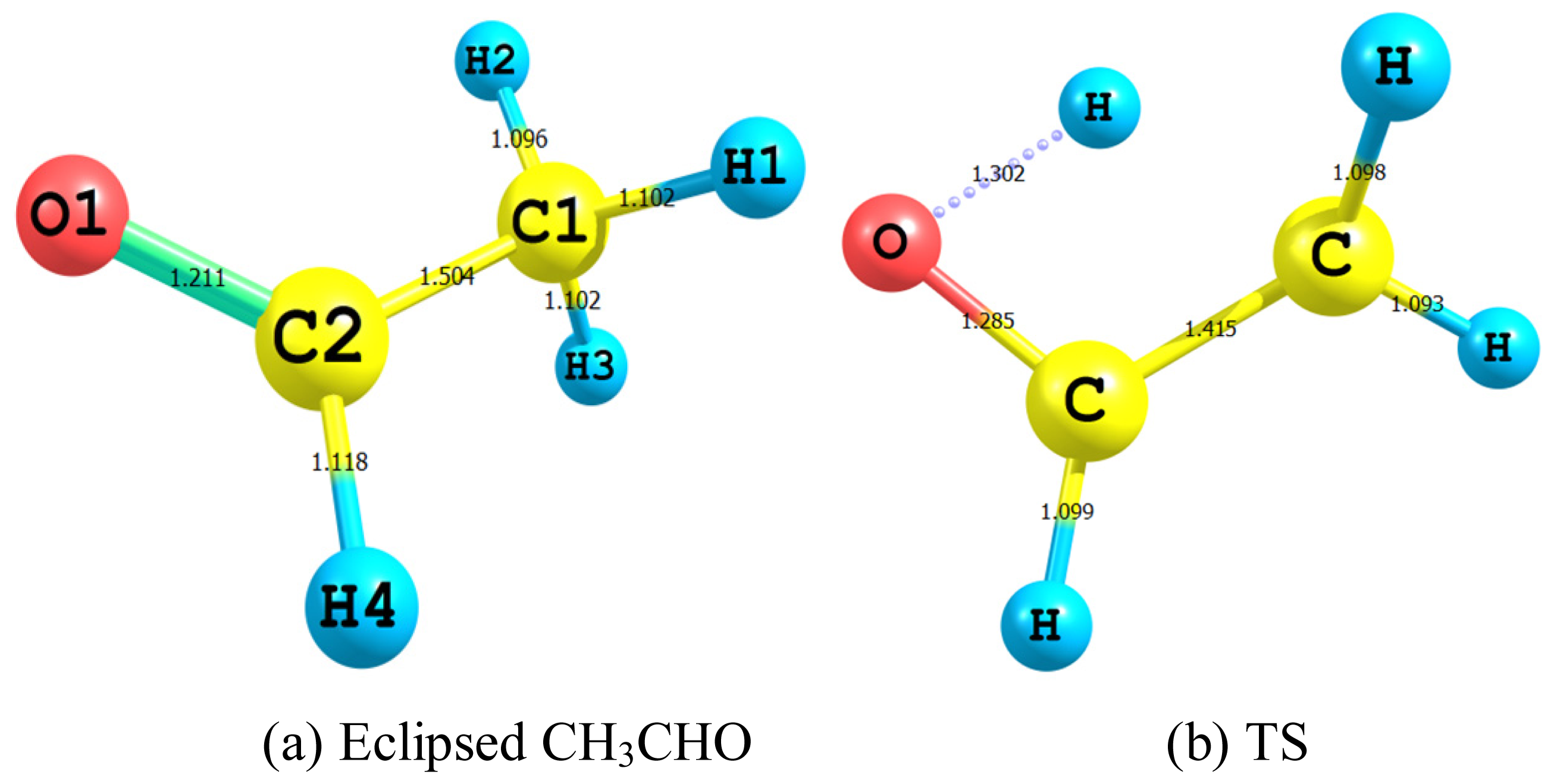
2.2.1. Geometry
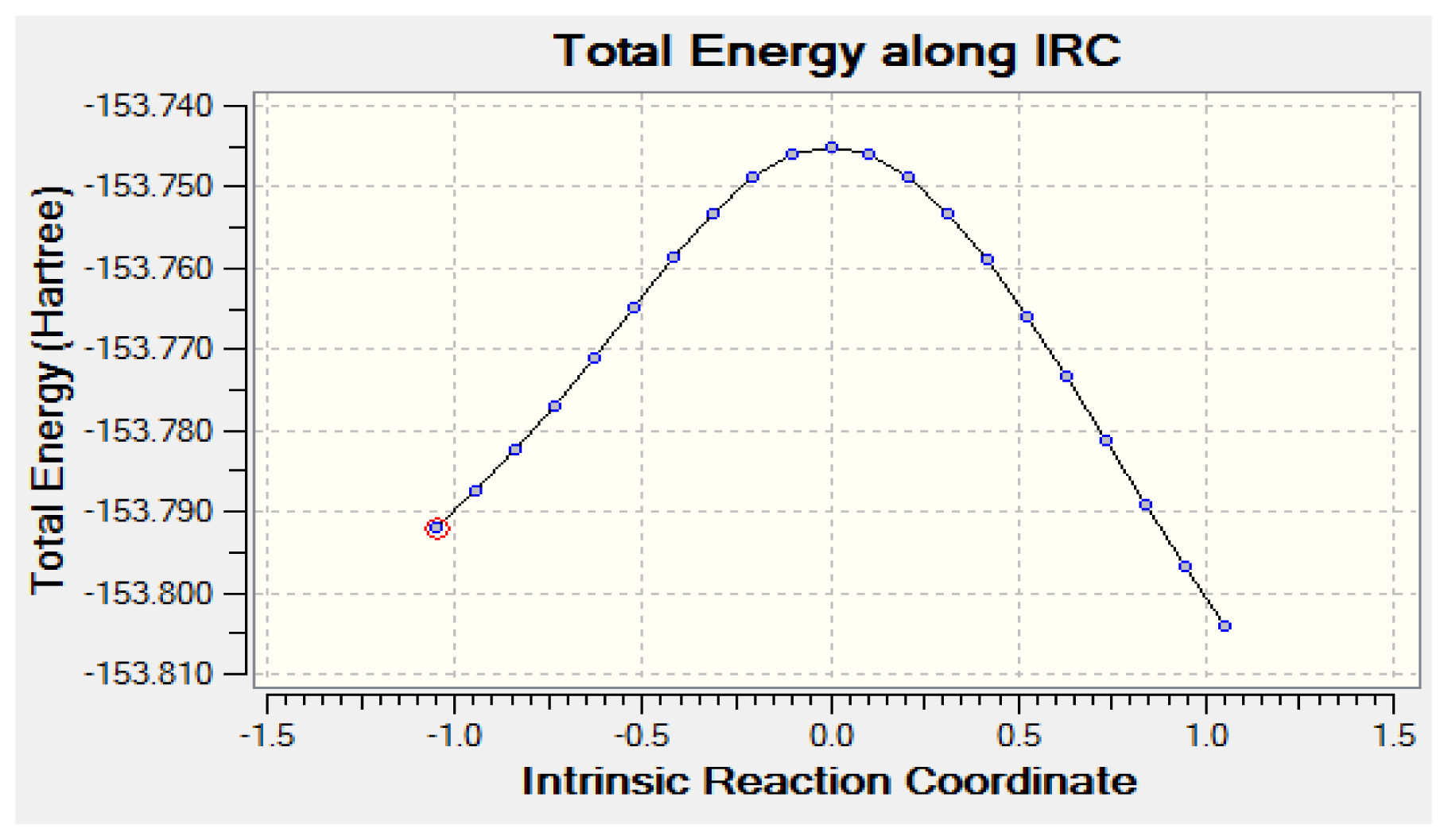
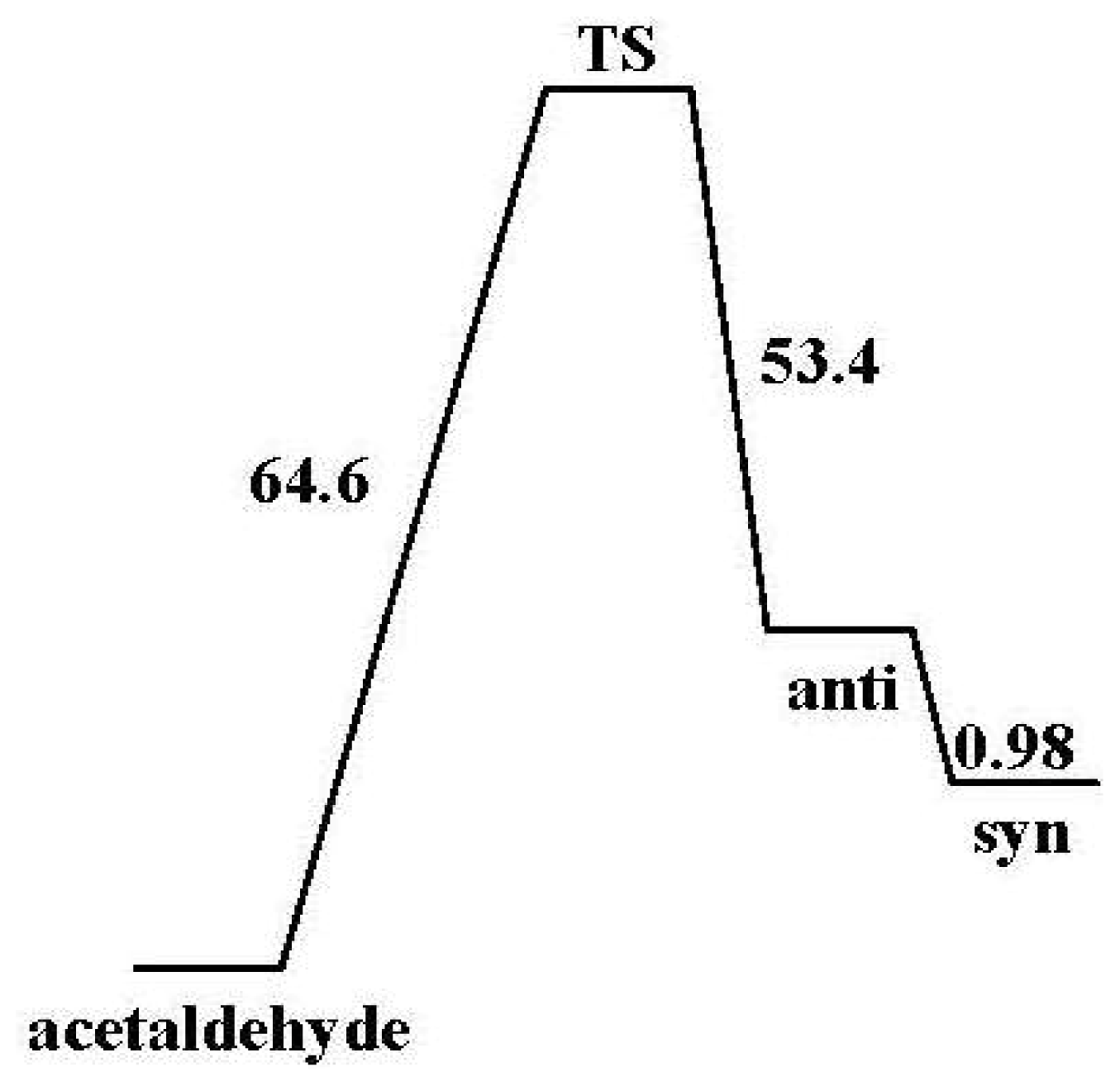
2.2.2. Activation Energies and Relative Stabilities
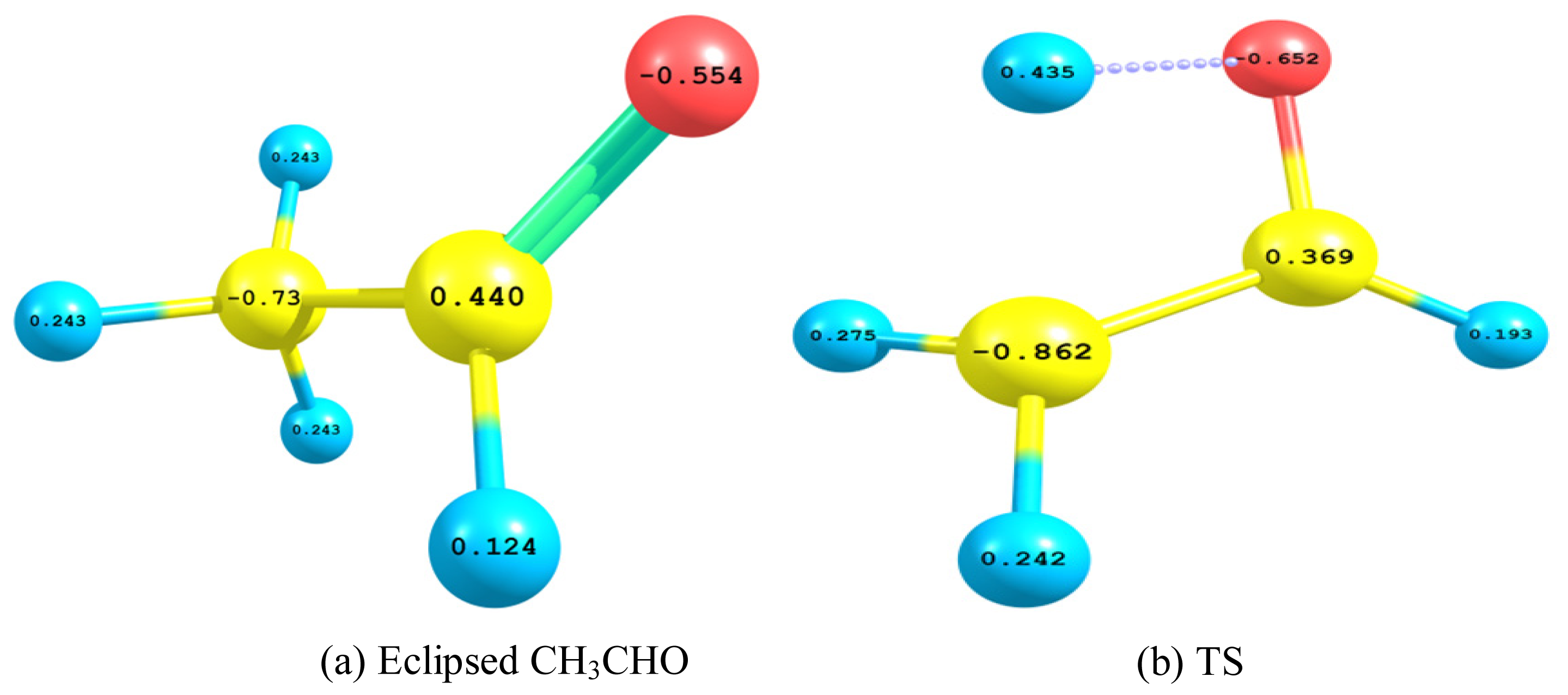
2.2.3. Natural Bond Orbital (NBO) Analysis
3. Computational Methods
4. Conclusion
- The MP2 and DFT/B3LYP methods with the 6-311++G(d,p) and aug-cc-pvdz basis sets have been used to probe (mainly through NBO calculations): (a) the origin of relative stability preference for the eclipsed acetaldehyde over its bisected counterpart and (b) the elucidation of the 1,3-proton shift mechanism of the tautomerization reaction of eclipsed acetaldehyde and vinyl alcohol.
- The elected model chemistries addressed the problems investigated in a consistent and complementary manner. It is apparent that the relative stability of eclipsed/bisected acetaldehyde conformers is both basis set and method dependent. In comparison to the aug-cc-pvdz basis set, the 6-311++G(d,p) gave lower comparable values (1.02 and 1.09 kcal/mol) at both MP2 and B3LYP methods. Thus, we anticipate that a modern technology experimental reexamination of eclipsed/bisected acetaldehyde would lead to a difference in stability in the order of 1.06 ± 0.04 kcal/mol. As such it will be in line with our lower limits of 1.02 and 1.09 kcal/mol.
- The chemistry models have overestimated the dipole moments of the acetaldehyde conformers (eclipsed form always has a higher value) in comparison to the experimental [16] value of 2.750 ± 0.006 Debye. The MP2/aug-cc-pvdz level proved to be superior over other tested chemistry levels in reproducing the dipole moments of eclipsed and bisected acetaldehyde of 5.5% and 1.7% higher, respectively, compared to the experimental value [16].
- The Lewis 2-electron localized structure favored the bisected conformer of acetaldehyde over that of the eclipsed one. Apparently this originated from the minimal steric hindrance. However, the overall preference for the eclipsed conformer arose mainly from the hyperconjugative interactions. In particular, the vicinal antiperiplanar (σC1–H2→σ*C2–H4 and σC2–H4→σ*C1–H2) interactions played the major role in the competiveness of the eclipsed conformer.
- The DFT/B3LYP method produced lower barrier heights than those estimated by MP2 model. In contrast, the aug-cc-pvdz basis set achieved the lowest activation energies of 64.61 kcal/mol.
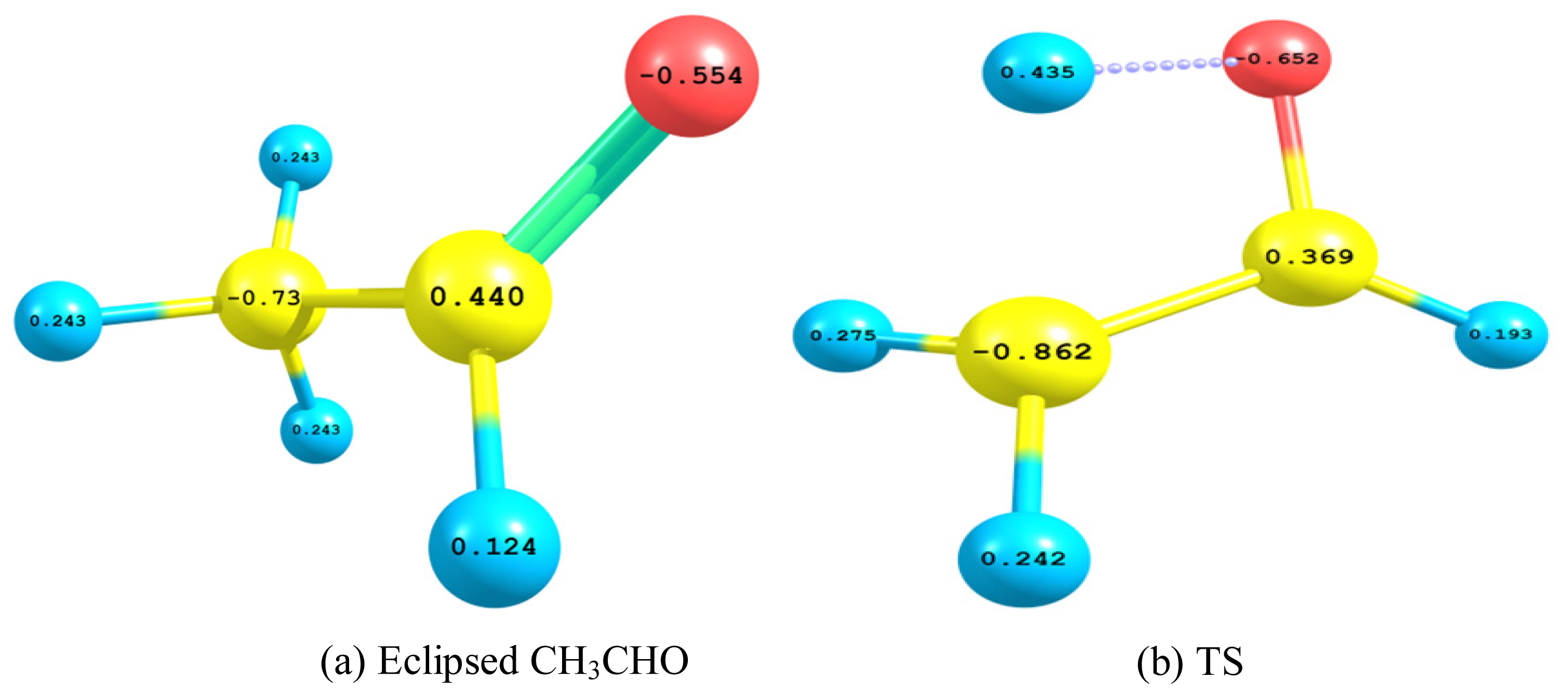
- The TS showed geometrical features that described the emergence of a four-membered ring species. This is achieved through an intramolecular migration of H2 accompanied by hybridization changes on heavy atoms. The NBO analysis of both eclipsed acetaldehyde and TS is in line with this intuition. The support for this prediction is based on the strong hyperconjugative interactions between the orbitals of the C1–H2 and C2–H4 bonds. This led to the weakness of the C1–H2 bond and a consequent intramolecular 1,3-proton shift between donor and acceptor sites.
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Hagemeyer, H.J. Encyclopedia of Chemical Technology; John Wiley & Sons: New York, NY, USA; p. 2001.
- Sowin, T.J.; Melcher, L.M. Encyclopedia of Reagents for Organic Synthesis, 2nd Ed; Paquette, L., Ed.; John Wiley & Sons: New York, NY, USA, 2004; Volume 14. [Google Scholar]
- Kilb, R.W.; Lin, C.C.; Wilson, E.B. Calculation of energy levels for internal torsion and over-all rotation. II. CH3CHO type molecules; acetaldehyde spectra. J. Chem. Phys 1957, 26, 1695–1703. [Google Scholar]
- Souter, C.E.; Wood, J.L. Combined infrared and microwave determination of torsional parameters. J. Chem. Phys 1970, 52, 674–682. [Google Scholar]
- Zanoun, A.; Durier, V.; Belaidi, A.; Vergoten, G. The SPASIBA force field of aldehydes. Part I: Structure and vibrational wavenumbers of methanal, ethanal and propanal. J. Mol. Struct 1999, 476, 261–270. [Google Scholar]
- Munoz-Caro, C.; Nino, A.; Moule, D.C. Definition of a periodic methyl torsional coordinate in the S0 state of acetaldehyde. J. Mol. Struct 1995, 350, 83–89. [Google Scholar]
- Eusuf, M.; Laidler, K.J. Kinetics and mechanisms of the thermal decomposition of acetaldehyde: I. The uninhibited reaction. Can. J. Chem 1964, 42, 1851–1860. [Google Scholar]
- Blank, B.; Fischer, H. Chemically Induced Dynamic Nuclear Polarization XI. Intermediary vinyl alcohol during photochemical reactions of acetaldehyde and of acetoin. Helv. Chim. Acta 1973, 56, 506–510. [Google Scholar]
- Vasiliou, A.; Piech, K.; Ellison, G.B.; Nimlos, M.R.; Daily, J.W.; Stanton, J.F. Thermal Decomposition of Acetaldehyde Studied by Matrix IR and Pims Spectroscopy. Proceedings of International Symposim on Molecular Spectroscopy, 65th Meeting, Ohio State University, Columbus, OH, USA, 21–25 June 2010.
- Vasiliou, A.; Piech, K.; Ellison, G.B.; Nimlos, M.R.; Daily, J.W.; Stanton, J.F. The Products of the Thermal Decomposition of CH3CHO. Proceedings of International Symposim on Molecular Spectroscopy, 66th Meeting, Ohio State University, Columbus, OH, USA, 20–24 June 2011.
- Holmes, J.H.; Lossing, F.P. Heats of formation of ionic and neutral enols of acetaldehyde and acetone. J. Am. Chem. Soc 1982, 104, 2648–2649. [Google Scholar]
- Smith, B.J.; Bouma, W.J.; Radom, L. Unimolecular rearrangements connecting hydroxyethylidene (CH3–C–OH), acetaldehyde (CH3–CH:O), and vinyl alcohol (CH2:CH–OH). J. Am. Chem. Soc 1991, 113, 6452–6458. [Google Scholar]
- Rosenfeld, R.N.; Weiner, B.R. Photofragmentation of acrylic acid and methacrylic acid in the gas phase. J. Am. Chem. Soc 1983, 105, 6233–6236. [Google Scholar]
- Wesdemiotis, C.; McLafferty, F.W. Hydroxyethylidene (CH3–C–OH), but not ethenol, tautomerizes to ethanol. J. Am. Chem. Soc. 1987, 109, 4760–4761. [Google Scholar]
- Andres, J.; Domingo, L.R.; Picher, M.T.; Safont, V.S. Comparative theoretical study of transition structures, barrier heights, and reaction energies for the intramolecular tautomerization in acetaldehyde/vinyl alcohol and acetaldimine/vinylamine systems. Int. J. Quant. Chem. 1998, 66, 9–24. [Google Scholar]
- Turner, P.H.; Cox, A.P. Microwave spectrum, structure, dipole moment and centrifugal distortion of nitrosomethane. Dipole moment of acetaldehyde. J. Chem. Soc. Faraday Trans. 2 1978, 74, 533–559. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev 1988, 88, 899–926. [Google Scholar]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree-Fock water dimer. J. Phys. Chem 1983, 78, 4066–4073. [Google Scholar]
- Wang, K.; Zhang, H.; Liu, Y. Ab initio study of hyperconjugative effect on electronic wavefunctions of 2-chloroethanol. Chin. J. Chem. Phys 2011, 24, 434–438. [Google Scholar]
- Yamamoto, T.; Kaneno, D.; Tomoda, S. The origin of cis effect in 1,2-dihaloethenes: The quantitative comparison of electron delocalizations and steric exchange repulsions. Bull. Chem. Soc. Jpn 2008, 81, 1415–1422. [Google Scholar]
- Kato, C.; Konaka, S.; Iijima, T.; Kimura, M. Electron diffraction studies of formaldehyde, acetaldehyde and acetone. Bull. Chem. Soc. Jpn 1969, 42, 2148–2150. [Google Scholar]
- Rodler, M.; Bauder, A. Structure of syn-vinyl alcohol determined by microwave spectroscopy. J. Am. Chem. Soc. 1984, 106, 4025–4028. [Google Scholar]
- Rodler, M. Microwave spectrum, dipole moment, and structure of anti-vinyl alcohol. J. Mol. Spectrosc 1985, 114, 23–30. [Google Scholar]
- Precomputed vibrational scaling factors. Available online: http://cccbdb.nist.gov/vibscalejust.asp accessed on 21 September 2012.
- Guo, D.; Goodman, L. Nature of barrier forces in acetaldehyde. J. Phys. Chem 1996, 100, 12540–12545. [Google Scholar]
- Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; Petersson, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc: Wallingford, CT, USA, 2009. [Google Scholar]
- Frisch, A.; Dennington, R.D.; Keith, T.A. GaussView Version 3.0; Gaussian Inc: Pittsburgh, PA, USA; p. 2003.
- Glendenning, E.D.; Reed, A.E.; Carpenter, J.E.; Weihold, F. NBO Version 3.1; Gaussian Inc: Pittsburgh, PA, USA; p. 1998.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MP2/6-311++G(d,p) | MP2/aug-cc-pvdz | B3LYP/6-311++G(d,p) | B3LYP/aug-cc-pvdz | |
|---|---|---|---|---|
| EFull Eclipsed | −153.44965823 | −153.41831870 | −153.88225230 | −153.85321084 |
| EFull Bisected | −153.44803394 | −153.41641243 | −153.88051576 | −153.85128459 |
| ΔET/kcal/mol | 1.02 | 1.19 | 1.09 | 1.21 |
| EL Eclipsed | - | - | −153.606588212 | −153.587544947 |
| EL Bisected | - | - | −153.613900827 | −153.595745153 |
| ΔEL/kcal/mol | 4.59 | 5.15 | ||
| Delocalization Energy/kcal/mol | 5.68 | 6.35 | ||
| Energy Gain = ΔET/kcal/mol | 1.09 | 1.20 | ||
| Exptl. a ΔET | 1.162 kcal/mol | |||
| μ/D Eclipsed | 2.943 | 2.907 | 3.376 | 3.360 |
| μ/D Bisected | 2.835 | 2.802 | 3.293 | 3.282 |
| Exptl. b μ/D | 2.750 ± 0.006 D | |||
| Interaction | Eclipsed | Sum | Interaction | Bisected | Sum |
|---|---|---|---|---|---|
| σC1–H1→σ*CO | 5.33 | 20.26 | σC1–H1→σ*CO | 4.51 | 17.56 |
| σC1–H1→π*CO | 2.02 | σC1–H2→σ*C2–H4 | 1.34 | ||
| σC1–H3→σ*CO | 5.33 | σC1–H2→σ*CO | 4.68 | ||
| σC1–H3→π*CO | 2.02 | σC1–H3→σ*C2–H4 | 1.34 | ||
| σC1–H2→σ*C2–H4 | 3.03 | σC1–H3→σ*CO | 4.69 | ||
| σC2–H4→σ*C1–H2 | 2.53 | - | - | ||
| nO1→σ*CC | 1.53 | 45.09 | nO1→σ*CC | 1.38 | 44.36 |
| nO1→σ*C2–H4 | 1.07 | nO1→σ*C2–H4 | 1.06 | ||
| nO2→σ*CC | 18.90 | nO2→σ*CC | 19.13 | ||
| nO2→σ*C2–H4 | 23.59 | nO2→σ*C2–H4 | 22.79 | ||
| Sum | 65.35 | 65.35 | Sum | 61.92 | 61.92 |
| Definition | CH3CHO | TS | Syn CH2CHOH | Anti CH2CHOH |
|---|---|---|---|---|
| C1–H1 | 1.102 (1.086) b | 1.093 | 1.088 (1.070) c | 1.087 (1.073) d |
| C1–H3 | 1.103 (1.086) | 1.098 | 1.092(1.079) | 1.089 (1.078) |
| C1–H2 | 1.096 (1.079) | 1.507 | - | - |
| C–C | 1.504 (1.501) | 1.415 | 1.337 (1.326) | 1.335 (1.315) |
| C2–H4 | 1.118 (1.114) | 1.099 | 1.090 (1.086) | 1.093 (1.075) |
| C–O | 1.212 (1.216) | 1.285 | 1.366 (1.372) | 1.372 (1.352) |
| OH | - | 1.302 | 0.967 (0.969) | 0.963 (0.941) |
| H1C1H3 | 109.57 (108.3) | 113.78 | 117.89 (118.8) | 118.85 (119.9) |
| H1C1C2 | 109.32 (109.2) | 122.22 | 119.74 (119.5) | 119.55 (119.9) |
| CCO | 124.71 (123.9) | 110.51 | 126.94 (126.2) | 122.15 (122.7) |
| H4C2O | 119.97 (117.5) | 119.12 | 110.47 (110.7) | 115.73 (115.7) |
| H1CCO | −121.85 | −152.96 | 180.00 | 180.00 |
| H3CCO | 121.85 | 66.50 | 0.00 | 0.00 |
| H2CCH4 | −179.99 | 168.17 | 180.00 | 180.00 |
| H2CCO | 0.002 | −9.09 | 180.00 | 180.00 |
| System | CH3CHO | TS | ΔE1 | SYNCH2CHOH | ΔE2 | ANTICH2CHOH |
|---|---|---|---|---|---|---|
| B3LYP/6-311++G(d,p) | −153.82889034 | −153.72316138 | −153.81118301 | −153.80933272 | ||
| Activation energy | 66.35 | - | 11.70 | 55.23 | 1.16 | 54.07 |
| B3LYP/aug-cc-pvdz | −153.7998880 | −153.69692622 | −153.78350726 | −153.78194342 | ||
| Activation energy | 64.61 | - | 10.71 | 54.33 | 0.98 | 53.35 |
| MP2/6-311++G(d,p) | −153.39641833 | −153.28889802 | −153.37687106 | −153.37501719 | ||
| Activation energy | 67.47 | - | 12.85 | 55.20 | 1.16 | 54.04 |
| MP2/aug-cc-pvdz | −153.36503858 | −153.26034014 | −153.34649926 | −153.34470879 | ||
| Activation energy | 65.70 | - | 12.20 | 54.07 | 1.13 | 52.94 |
| Interaction/Compound | σC1H1 →σ*CO | σC1H3 →σ*CO | σC1H2 →σ*C2H4 | σC2H4 →σ*C1H2 | nO2 →σ*CC | nO2 →σ*C2H4 | nO2 →σ*C1H2 |
|---|---|---|---|---|---|---|---|
| CH3CHO | 5.33 | 5.33 | 3.03 | 2.53 | 18.90 | 23.59 | - |
| TS | 9.50 | 2.37 | 30.26 | 2.53 | 3.56 | 10.53 | 96.08 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Osman, O.I.; Alyoubi, A.O.; Elroby, S.A.K.; Hilal, R.H.; Aziz, S.G. Eclipsed Acetaldehyde as a Precursor for Producing Vinyl Alcohol. Int. J. Mol. Sci. 2012, 13, 15360-15372. https://doi.org/10.3390/ijms131115360
Osman OI, Alyoubi AO, Elroby SAK, Hilal RH, Aziz SG. Eclipsed Acetaldehyde as a Precursor for Producing Vinyl Alcohol. International Journal of Molecular Sciences. 2012; 13(11):15360-15372. https://doi.org/10.3390/ijms131115360
Chicago/Turabian StyleOsman, Osman I., Abdulrahman O. Alyoubi, Shabaan A. K. Elroby, Rifaat H. Hilal, and Saadullah G. Aziz. 2012. "Eclipsed Acetaldehyde as a Precursor for Producing Vinyl Alcohol" International Journal of Molecular Sciences 13, no. 11: 15360-15372. https://doi.org/10.3390/ijms131115360
APA StyleOsman, O. I., Alyoubi, A. O., Elroby, S. A. K., Hilal, R. H., & Aziz, S. G. (2012). Eclipsed Acetaldehyde as a Precursor for Producing Vinyl Alcohol. International Journal of Molecular Sciences, 13(11), 15360-15372. https://doi.org/10.3390/ijms131115360