Highly Regio- and Stereoselective Diels-Alder Cycloadditions via Two-Step and Multicomponent Reactions Promoted by Infrared Irradiation under Solvent-Free Conditions

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
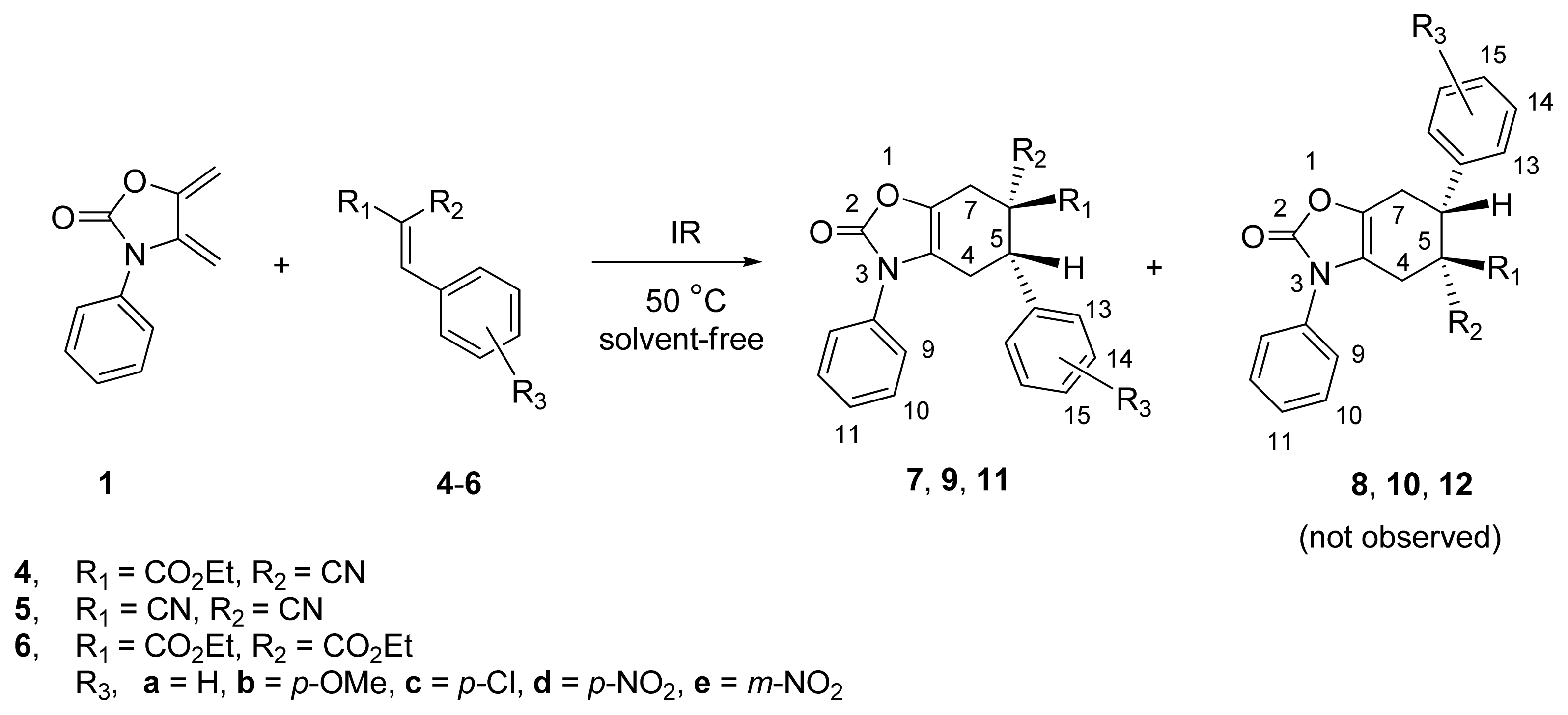
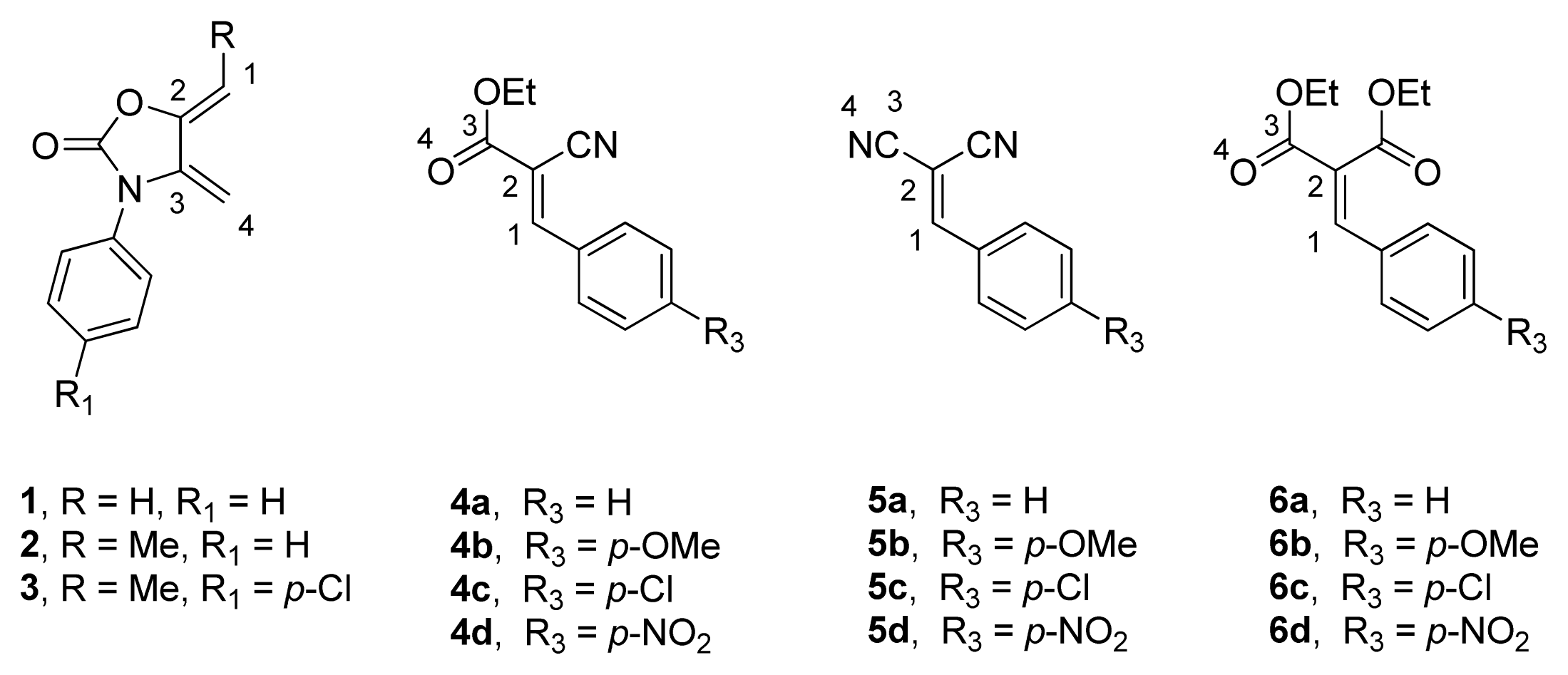
2.1. Diels-Alder Cycloaddition with Diene 1
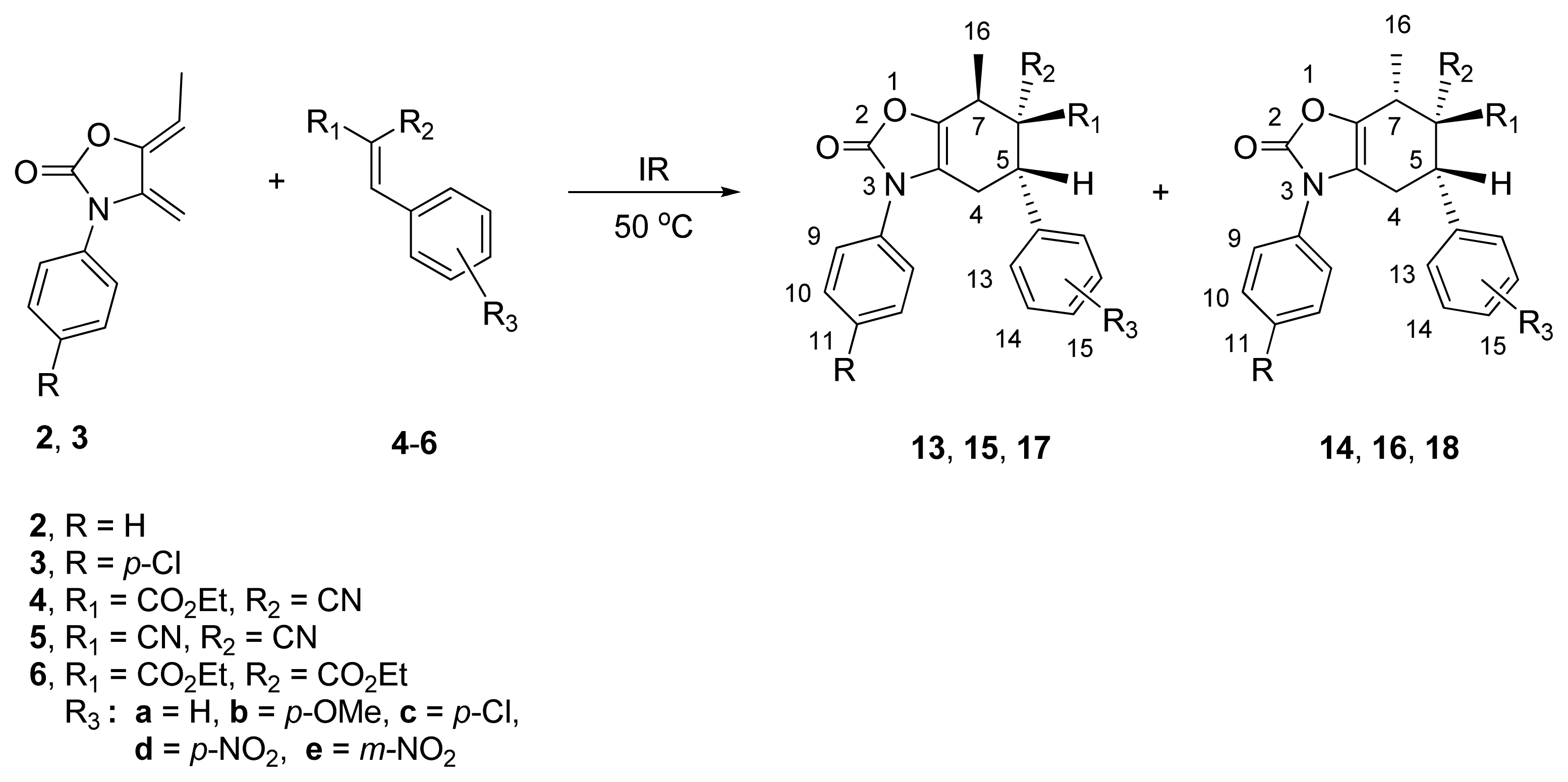
2.2. Diels-Alder Cycloaddition with Dienes 2 and 3
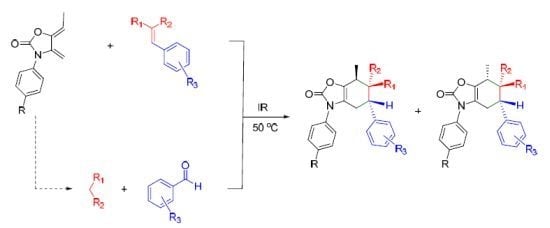
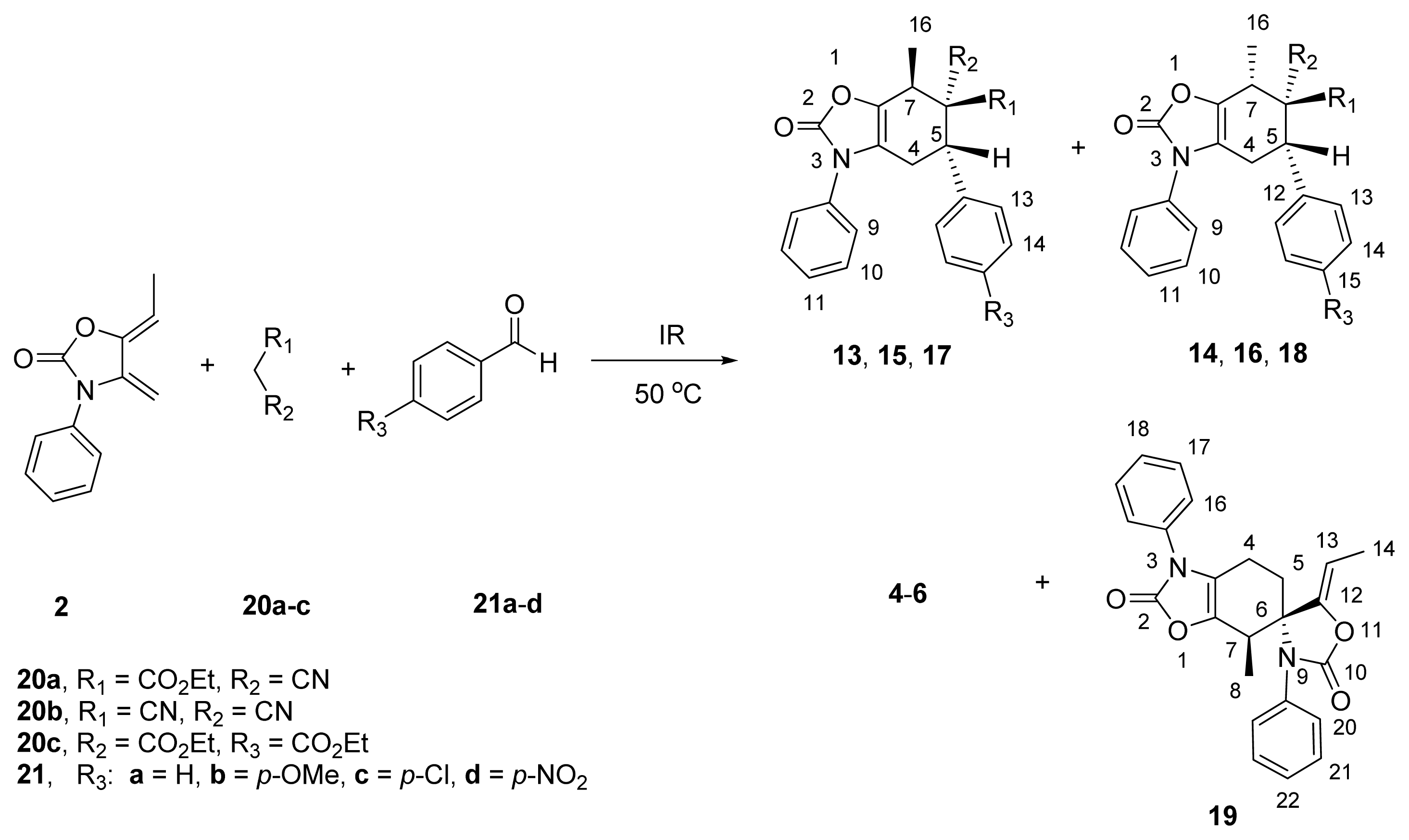
2.3. Multicomponent Reactions
2.4. Diels-Alder Regioselectivity and FMO Theory
3. Experimental Section
3.1. General Procedures and Instrumentation
3.2. General Procedures for the Synthesis of Adducts 7a–e, 9a–d, 11a–c, 13a–e/14a–e, 15a–e/16a–e and 17a–c via a Two-Step Reaction. Method A
3.3. General Procedure for the Synthesis of Adducts 13a–d/14a–d and 15a–d/16a–d via a One-Step Reaction. Method B
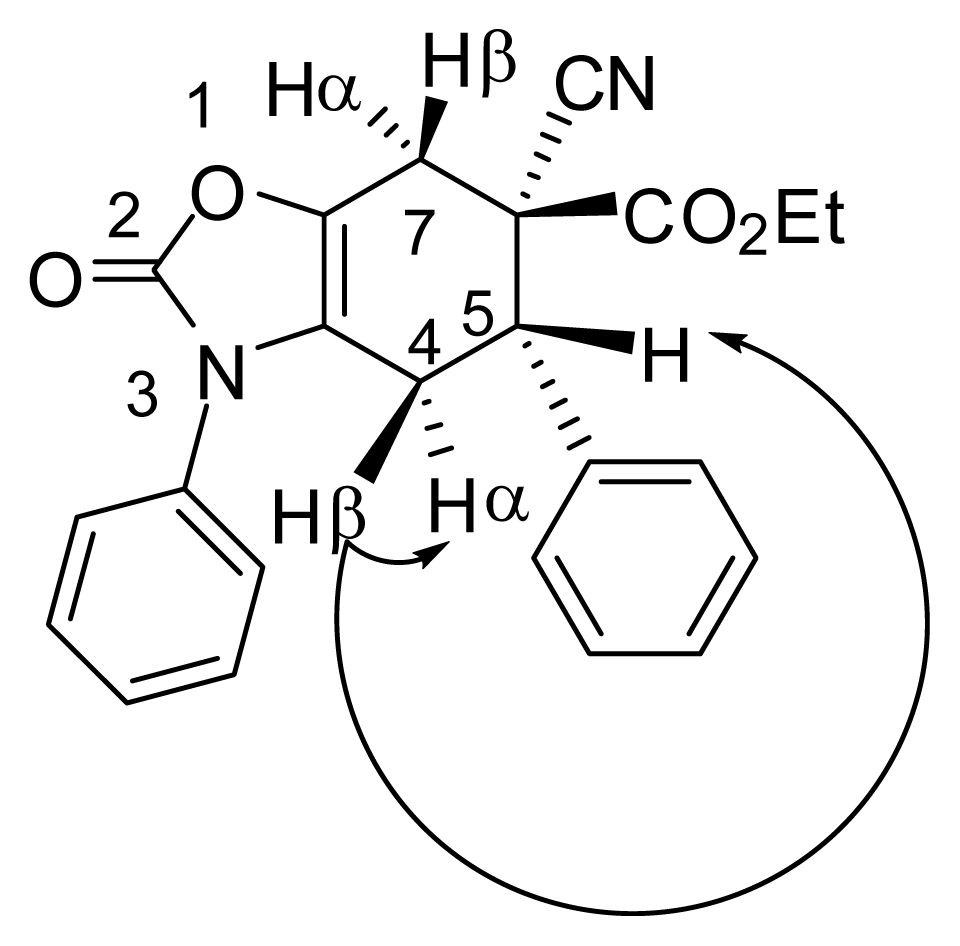
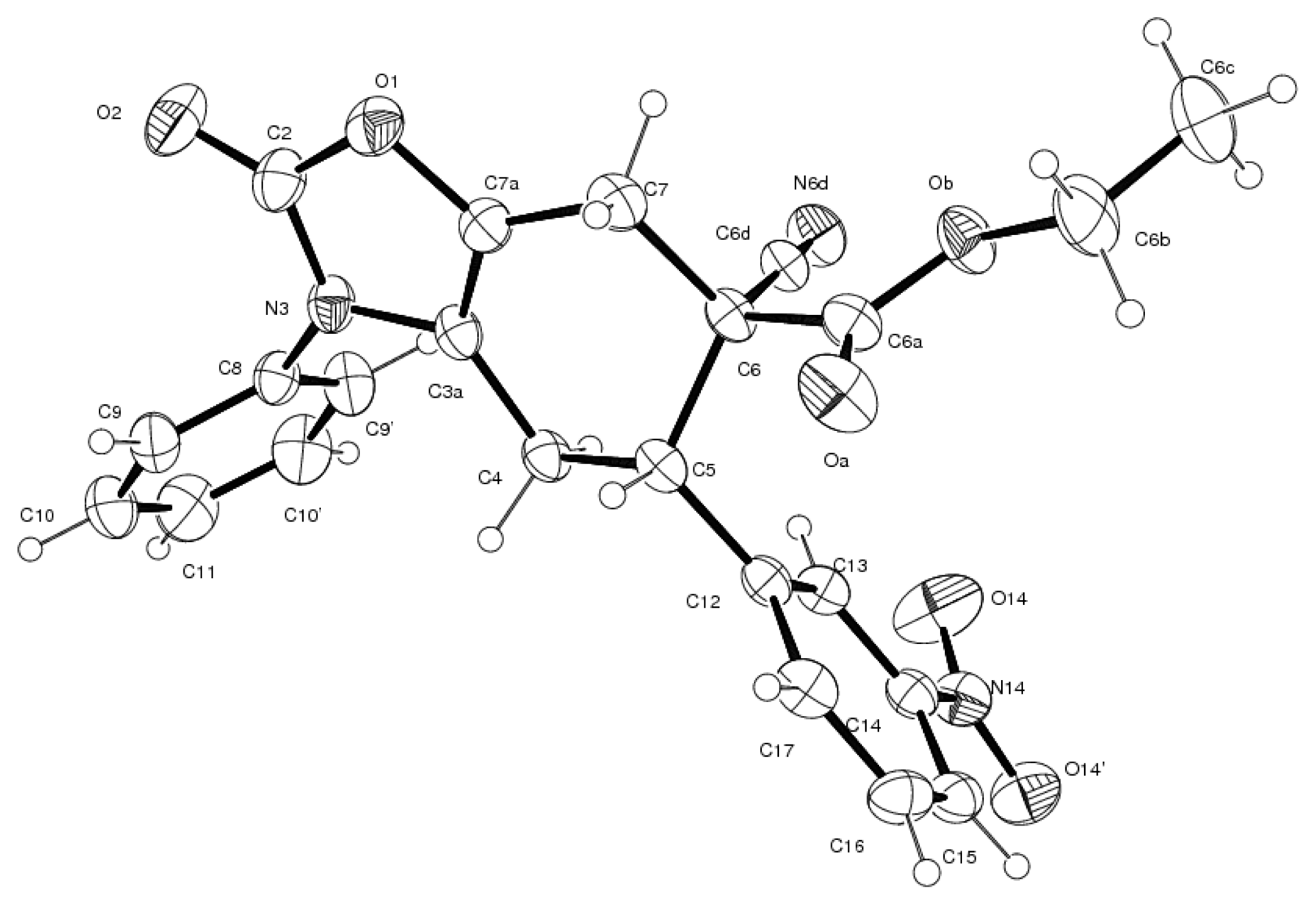
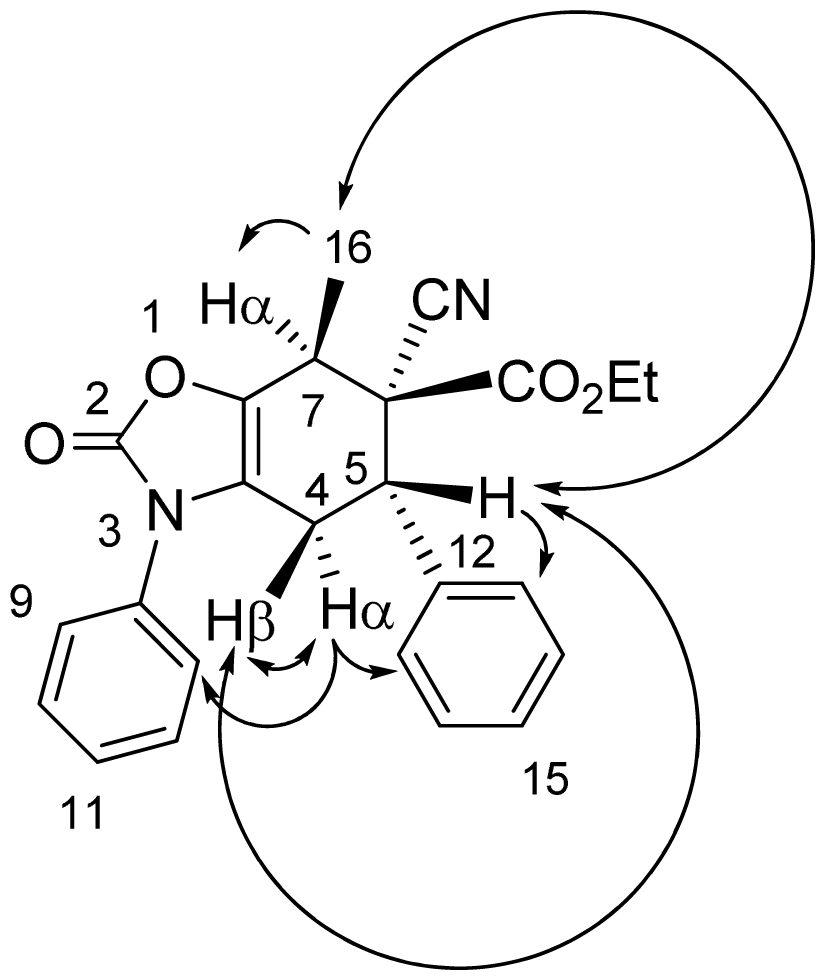
3.4. X-ray Structure Study of 7e, 13a and 19
3.5. Theoretical Calculations
4. Conclusions
Supporting Information
ijms-13-02590-s001.pdfAcknowledgments
- Supporting Information AvailableTables summarizing 1H and 13C NMR data of the adducts 7a–e, 9a–d, 11a–d, 13a–e, 14a–e, 15a–e, 16a–e and 17a–d including images of IR, 1H and 13C NMR (HMQC, HMBC and NOE experiments) and mass spectra for most of the products. Cartesian Coordinates (B3LYP/6-31G**), energies, and lowest vibrational frequencies (RHF/6-31G**) of the optimized geometries of diene 3 and dienophiles 4a–d, 5a–d and 6a–d. Crystallographic information for 7e, 13a and 19 in CIF format, including X-ray diffraction data, atomic coordinates, thermal parameters and complete bond distances and angles. This material is available free of charge via the Internet at the Cambridge Crystallographic Data Centre (e-mail: deposit@ccdc.cam.ac.uk) as supplementary publication: CCDC 756200 (7e), CCDC 756201 (13a), and CCDC 829554 (19).
References
- Sabot, C.; Oueis, E.; Brune, X.; Renard, P.-Y. Synthesis of polisubstituted 3-hydroxypyridines via the revisited hetero-Diels-Alder reaction of 5-alkoxyoxazoles whit dienophiles. Chem. Commun 2012, 48, 768–770. [Google Scholar]
- Suárez-Moreno, G.V.; González-Zamora, E.; Méndez, F. Oxazole as an electron-deficient diene in the Diels-Alder reaction. Org. Lett 2011, 13, 6358–6361. [Google Scholar]
- Nawrat, C.C.; Lewis, W.; Moody, C.J. Synthesis of amino-1,4-benzoquinones and their use in Diels-Alder approaches to the aminonaphthoquinone antibiotics. J. Org. Chem 2011, 76, 7872–7881. [Google Scholar]
- Martin, N; Seoane, C.; Hanack, M. Recent advanced in o-quinodimethane chemistry. Org. Prep. Proc. Int. 1991, 23, 237–272. [Google Scholar]
- Charlton, J.L.; Alauddin, M.M. Orthoquinodimethanes. Tetrahedron 1987, 43, 2873–2889. [Google Scholar]
- Fringuelli, F.; Taticchi, A. The Diels-Alder Reaction Selected Practical Methods; John Wiley: New York, NY, USA, 2002. [Google Scholar]
- Sabitha, G.; Reddy, G.S.; Kiran, K.; Rajkumar, M.; Yadav, J.S.; Ramakrishna, K.V.S.; Kunwar, A.C. Iodotrimethysilane induced diastereoselective synthesis of tetrahydropyranones by a tandem Knoevenagel-Michael reaction. Tetrahedron Lett 2003, 44, 7455–7457. [Google Scholar]
- Ramachary, D.B.; Barbas, C. Towards organo-click chemistry. Development of organocatalytic multicomponent reactions through combinations of Aldol, Wittig, Knoevenagel, Michael, Diels-Alder and Huisgen cycloaddition reactions. Chem. Eur. J 2004, 10, 5323–5331. [Google Scholar]
- Palasz, A.; Palasz, T. Knoevenagel condensation of cyclic ketones with benzoylacetonitrile and N,N'-dimethylbarbituric acid. Application of sterically hindered condensation products in the synthesis of spiro and dispiropyrans by hetero-Diels-Alder reactions. Tetrahedron 2011, 67, 1422–1431. [Google Scholar]
- Kuttruff, C.A.; Zipse, H.; Trauner, D. Concise total syntheses of variecolortides A and B through an unusual Hetero-Diels-Alder reaction. Angew. Chem. Chem. Int. Ed 2011, 50, 1402–1405. [Google Scholar]
- Kim, I.; Kim, S.G.; Choi, J.; Lee, G.H. Facile synthesis of benzo-fused 2,8-dioxabiclyclo [3.3.1]nonane derivatives via a domino Knoevenagel condensation/hetero-Diels-Alder reaction sequence. Tetrahedron 2008, 64, 664–671. [Google Scholar]
- Pizzirani, D.; Roberti, M.; Recanatini, M. Domino Knoevenagel/Diels-Alder sequence coupled to Suzuki reaction: A valuable synthethic platform for chemical biology. Tetrahedron Lett 2007, 48, 7120–7124. [Google Scholar]
- Amantini, D.; Fringuelli, F.; Piermatti, O.; Pizzo, F.; Vaccaro, L. Water, a clean, inexpensive, and re-usable reaction medium. One-pot synthesis of (E)-2-aryl-1-cyano-1-nitroethenes. Green Chem 2001, 3, 229–232. [Google Scholar]
- Fernandez, I.; Dyker, C.A.; DeHope, A.; Donnadieu, B.; Frenking, G.; Bertrand, G. Exocyclic delocalization at the expense of aromaticity in 3,5-bis(π-donor) substituted pirazolium ions and corresponding cyclic bent allene. J. Amer. Chem. Soc 2009, 131, 11875–11881. [Google Scholar]
- Sikervar, V.; Fuchs, P.L. SN2' addition/1,2-elimination of dimethylsulfonium methilide with epoxy vinyl sulfones: Synthesis of exocyclic cross-conjugated dienyl sulfones. Chem. Commun 2011, 47, 3472–3474. [Google Scholar]
- Hernández, R.; Sánchez, J.M.; Gómez, A.; Trujillo, G.; Aboytes, R.; Zepeda, G.; Bates, R.W.; Tamariz, J. Novel heterocyclic outer-ring dienes: N-alkyl- and N-aryl substituted 4,5-dimethylene-2-oxazolidinones. Heterocycles 1993, 36, 1951–1956. [Google Scholar]
- Mandal, A.B.; Gómez, A.; Trujillo, G.; Méndez, F.; Jiménez, H.A.; Rosales, M.J.; Martínez, R.; Delgado, F.; Tamariz, J. One-step synthesis and highly regio- and stereoselective Diels-Alder of novel exo-2-oxazolidinone dienes. J. Org. Chem 1997, 62, 4105–4115. [Google Scholar]
- Fuentes, A.; Martínez-Palou, R.; Jiménez-Vázquez, H.A.; Delgado, F.; Reyes, A.; Tamariz, J. Diels-Alder reactions of 2-oxazolidinone dienes in polar solvents using catalysis or non-conventional energy sources. Monatsh. Chem 2005, 136, 177–192. [Google Scholar]
- Martínez, R.; Jiménez-Vázquez, H.A.; Reyes, A.; Tamariz, J. Stereoselective synthesis of 4,5-diethylidene-oxazolidinones as new dienes in Diels-Alder reactions. Helv. Chim. Acta 2002, 85, 464–482. [Google Scholar]
- Martínez, R.; Jiménez-Vázquez, H.A.; Delgado, F.; Tamariz, J. Synthesis and highly Diels-Alder cycloadditions of the new dienes N-susbtituted 2,3,5,6-tetrahydrobenzoxazol-2-ones. Tetrahedron 2003, 59, 481–492. [Google Scholar]
- Benavides, A.; Peralta, J.; Delgado, F.; Tamariz, J. Total synthesis of the natural carbazoles murrayanine and murrayafoline A, based on the regioselective Diels-Alder addition of exo-2-oxazolidinone dienes. Synthesis 2004, 2499–2504. [Google Scholar]
- Bernal, P.; Benavides, A.; Bautista, R.; Tamariz, J. Exo-2-oxazolidinone dienes in the total synthesis of the natural carbazoles, 6-methoxymurrayanine and clausenine. Synthesis 2007, 1943–1948. [Google Scholar]
- Bernal, P.; Tamariz, J. Total synthesis of murrayanine involving 4,5-dimethyleneoxazolidin-2-ones and a palladium(0)-catalyzed diaryl insertion. Helv. Chim. Acta 2007, 90, 1449–1454. [Google Scholar]
- Bautista, R.; Bernal, P.; Montiel, L.E.; Delgado, F.; Tamariz, J. Total synthesis of the natural carbazoles glycozolicine, mukoline, and mukolidine, startingfrom 4,5-dimethyleneoxazolidin-2-ones. Synthesis 2011, 929–933. [Google Scholar]
- Reyes, L.; Mendoza, H.; Vázquez, M.A.; Ortega-Jiménez, F.; Fuentes-Benítes, A.; Jiménez-Vázquez, H.; Flores-Conde, M.I.; Miranda, R.; Tamariz, J.; Delgado, F. Synthesis of new polycyclic oxazol-2-one derivatives by a tandem [4+2] cycloaddition/cyclopentannulation/ 1,5-sigmatropic rearrangement process of Fischer (arylalkynyl)(alkoxy)carbenes and exo-2-oxazolidinone dienes. Organometallics 2008, 27, 4334–4345. [Google Scholar]
- Ortega-Jiménez, F.; Benavides, A.; Delgado, F.; Jiménez-Vázquez, H.A.; Tamariz, J. Synthesis and reactivity of η4-Diene-Fe(CO)3 complexes from exo-2-oxazolidinone dienes. A facile generation of stable conjugated enol-enamido species. Organometallics 2010, 29, 149–159. [Google Scholar]
- Delgado, F.; Tamariz, J.; Zepeda, G.; Landa, M.; Miranda, R.; García, J. Knoevenagel condensation catalyzed by a mexican bentonite using infrared irradiation. Synth. Commun 1995, 25, 753–759. [Google Scholar]
- Obrador, E.; Castro, M.; Tamariz, J.; Zepeda, G.; Miranda, R.; Delgado, F. Knovenagel condensation in heterogeneous phase catalyzed by IR radiation and tonsil actisil FF. Synth. Commun 1998, 28, 4649–4663. [Google Scholar]
- Alcerreca, G.; Sanabria, R.; Miranda, R.; Arroyo, G.; Tamariz, J.; Delgado, F. Preparation of benzylidene barbituric acids promoted by infrared irradiation, in the absence of solvent. Synth. Commun 2000, 30, 1295–1301. [Google Scholar]
- Penieres, G.; Miranda, R.; García, J.; Aceves, J.; Delgado, F. Modification of the Fischer indole synthesis. Heterocycl. Commun 1996, 2, 401–402. [Google Scholar]
- Osnaya, R.; Arroyo, G.; Parada, L.; Delgado, F.; Trujillo, J.; Salmón, S.; Miranda, R. Biginelli vs Hantzsch esters study under infrared radiation and solventless conditions. Arkivoc 2003, xi, 112–117. [Google Scholar]
- Martínez, J.; Velasco-Bejarano, B.; Delgado, F.; Pozas, R.; Torres Domínguez, H.M.; Trujillo, J.; Arroyo, G.A.; Miranda, R. Eco-contribution to the chemistry of perezone, a comparative study, using different modes of activation and solventless conditions. Nat. Prod. Commun 2008, 3, 1465–1468. [Google Scholar]
- Pool, G.C.; Teuben, J.H. IR Radiation as a Heat Source in Vacuum Sublimation. In Practical Organometallic Chemistry; Wayda, A.L., Darensbourg, M.Y.W., Eds.; Symposium Series, Washington DC, USA, 1987; Volume 357, pp. 30–33. [Google Scholar]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; John Wiley & Sons: Chichester, UK, 1976. [Google Scholar]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry. Reactions, Mechanisms, and Structure, 5th ed.; John Wiley & Sons, Inc: New York, NY, USA, 2001; p. 370. [Google Scholar]
- Argile, A.; Ruasse, M.-F. Reactivity and selectivity control by reactants and products. A general relationship between the selectivity and the position of the transition state. Tetrahedron Lett 1980, 21, 1327–1330. [Google Scholar]
- Bowden, K.; Stewart, R. Strongly basic systems—V:H-acidity scale based on the ionization of carbon acids. Tetrahedron 1965, 21, 261–266. [Google Scholar]
- Pearson, R.G.; Dillon, R.L. Rates of ionization of pseudo acids. Relation between rates and equilibria. J. Am. Chem. Soc 1953, 75, 2439–2443. [Google Scholar]
- Bell, R.P. The Proton in Chemistry; Cornell University Press: Ithaca, NY, USA, 1959. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Savetti correlational-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Gaussian 03, revision E.01; Gaussian Inc: Wallingford, CT, USA, 2004.
- Fukui, K. Recognition of stereochemical paths by orbital interaction. Accounts Chem. Res 1971, 4, 57–64. [Google Scholar]
- Eisenstein, O.; Lefour, J.M.; Anh, N.T.; Hudson, R.F. Simple prediction of cycloaddition orientation. I. Diels-Alder reactions. Tetrahedron 1977, 33, 523–531. [Google Scholar]
- Houk, K.N. Generalized frontier orbitals of alkenes and dienes. Regioselectivity in Diels-Alder reactions. J. Am. Chem. Soc 1973, 95, 4092–4094. [Google Scholar]
- Sustmann, R. Orbital energy control of cycloaddition reactivity. Pure Appl. Chem 1974, 40, 569–593. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr 2008, A64, 112–122. [Google Scholar]
- SHELX97, release 97-2; programs for crystal structure analysis; Institüt für Anorganische Chemie der Universität: Göttingen, Germany, 1997.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and refinement of crystal structures with SIR92. J. Appl. Crystallogr 1993, 26, 343–350. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr 1999, 32, 837–838. [Google Scholar]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr 2003, 36, 7–13. [Google Scholar]
- Farrugia, L.J. ORTEP-3 for Windows-a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr 1997, 30, 565. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Dienophile | R1 | R2 | R3 | Reaction Time (h) | Product e (%) |
| 1 | 4a | CO2Et | CN | H | 3.5 | 7a (73) |
| 2 b | 4a | CO2Et | CN | H | 20 | 7a (30) |
| 3 c | 4a | CO2Et | CN | H | 24 | 7a (20) |
| 4 d | 4a | CO2Et | CN | H | 24 | 7a (20) |
| 5 | 4b | CO2Et | CN | p-OMe | 4.0 | 7b (50) |
| 6 | 4c | CO2Et | CN | p-Cl | 3.5 | 7c (60) |
| 7 | 4d | CO2Et | CN | p-NO2 | 3.0 | 7d (80) |
| 8 | 4e | CO2Et | CN | m-NO2 | 3.5 | 7e (55) |
| 9 | 5a | CN | CN | H | 4.0 | 9a (80) |
| 10 | 5b | CN | CN | p-OMe | 4.5 | 9b (55) |
| 11 | 5c | CN | CN | p-Cl | 3.0 | 9c (75) |
| 12 | 5d | CN | CN | p-NO2 | 3.0 | 9d (85) |
| 13 | 6a | CO2Et | CO2Et | H | 5.0 | 11a (35) |
| 14 | 6b | CO2Et | CO2Et | p-OMe | 6.0 | 11b (25) |
| 15 | 6c | CO2Et | CO2Et | p-Cl | 5.0 | 11c (30) |
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Diene | Dienophile | R | R1 | R2 | R3 | Reaction Time (h) | Products (endo/exo) b | Yield c (%) |
| 1 | 2 | 4a | H | CO2Et | CN | H | 4.0 | 13a/14a (80:20) | 56/32 |
| 2 | 2 | 4b | H | CO2Et | CN | p-OMe | 5.0 | 13b/14b (75:25) | 65 d |
| 3 | 2 | 4c | H | CO2Et | CN | p-Cl | 4.5 | 13c/14c (68:32) | 60 d |
| 4 | 2 | 4d | H | CO2Et | CN | p-NO2 | 4.0 | 13d/14d (75:25) | 64 d |
| 5 | 3 | 4e | p-Cl | CO2Et | CN | m-NO2 | 4.5 | 13e/14e (75:25) | 70 d |
| 6 | 2 | 5a | H | CN | CN | H | 3.0 | 15a/16a (80:20) | 70 d |
| 7 | 2 | 5b | H | CN | CN | p-OMe | 4.0 | 15b/16b (82:18) | 55 d |
| 8 | 2 | 5c | H | CN | CN | p-Cl | 5.0 | 15c/16c (90:10) | 75 d |
| 9 | 2 | 5d | H | CN | CN | p-NO2 | 2.0 | 15d/16d (80:20) | 75/15 |
| 10 | 3 | 5b | p-Cl | CN | CN | p-OMe | 3.0 | 15e/16e (75:25) | 70/15 |
| 11 | 2 | 6a | H | CO2Et | CO2Et | H | 6.0 | 17a/18a (100:0) | 23 d |
| 12 | 2 | 6b | H | CO2Et | CO2Et | p-OMe | 5.0 | 17b/18b (100:0) | 32 d |
| 13 | 2 | 6c | H | CO2Et | CO2Et | p-Cl | 4.0 | 17c/18c (100:0) | 25 d |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Methylene Active | Benzaldehyde | Reaction Time (min) | By-Products (%) b | Adducts (endo/exo) c | Yield d (%) |
| 1 | 20a | 21a | 35 | 4a/19 (40:5) | 13a/14a (65:35) | 43/12 |
| 2 | 20a | 21b | 50 | 4b/19 (52:8) | 13b/14b (75:25) | 40 e |
| 3 | 20a | 21c | 30 | 4c/19 (35:5) | 13c/14c (68:32) | 60 e |
| 4 | 20a | 21d | 40 | 4d/19 (32:4) | 13d/14d (75:25) | 64 e |
| 5 | 20b | 21a | 30 | 5a/19 (40:5) | 15a/16a (70:30) | 55 e |
| 6 | 20b | 21b | 40 | 5b/19 (40:10) | 15b/16b (85:15) | 50 e |
| 7 | 20b | 21c | 30 | 5c/19 (20:5) | 15c/16c (80:20) | 65/10 |
| 8 | 20b | 21d | 35 | 5d/19 (25:5) | 15d/16d (70:30) | 55/15 |
| 9 | 20c | 21a | 150 | 6a/19 (64:36) | -- | -- |
| 10 | 20c | 21b | 210 | 6b/19 (70:30) | -- | -- |
| 11 | 20c | 21c | 240 | 6c/19 (60:40) | -- | -- |
| 12 | 20c | 21d | 240 | 6d/19 (65:35) | -- | -- |
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | |||||||||||
| Compd b | E (eV) | C1 | C2 | C3 | C4 | ΔCic | E (eV) | C1 | C2 | C3 | C4 | ΔCic) |
| 1d | −8.8051 | 0.246 | 0.164 | −0.209 | −0.326 | 0.080 | 2.9065 | 0.263 | −0.245 | −0.245 | 0.258 | −0.005 |
| 2d | −8.5610 | −0.257 | −0.199 | 0.198 | 0.320 | 0.063 | 3.1035 | 0.274 | −0.222 | −0.245 | 0.248 | −0.026 |
| 3 | −8.6408 | −0.277 | −0.220 | 0.199 | 0.324 | 0.047 | 2.7244 | 0.288 | −0.232 | −0.247 | 0.258 | −0.030 |
| 4a | −8.9382 | 0.122 | 0.276 | 0.015 | −0.114 | −0.154 | 1.0104 | 0.296 | −0.210 | −0.131 | 0.113 | 0.086 |
| 4b | −8.4299 | −0.084 | −0.260 | −0.020 | 0.105 | −0.176 | 1.2204 | 0.306 | −0.203 | −0.134 | 0.112 | 0.103 |
| 4c | −9.0541 | 0.116 | 0.265 | 0.014 | −0.109 | −0.149 | 0.7532 | 0.288 | −0.211 | −0.126 | 0.110 | 0.077 |
| 4d | −9.7679 | 0.160 | 0.284 | 0.008 | −0.121 | −0.124 | −0.1056 | 0.219 | −0.196 | −0.096 | 0.092 | 0.023 |
| 5a | −9.2234 | 0.126 | 0.275 | −0.058 | −0.149 | 0.5331 | 0.305 | −0.227 | −0.071 | 0.078 | ||
| 5b | −8.6859 | −0.087 | −0.261 | 0.044 | −0.174 | 0.7611 | 0.315 | −0.219 | −0.073 | 0.096 | ||
| 5c | −9.3227 | 0.118 | 0.263 | −0.055 | −0.145 | 0.2759 | 0.298 | −0.227 | −0.068 | 0.071 | ||
| 5d | −10.0495 | 0.163 | 0.280 | −0.073 | −0.117 | −0.5323 | 0.238 | −0.214 | −0.050 | 0.024 | ||
| 5c | −9.3227 | 0.118 | 0.263 | −0.055 | −0.145 | 0.2759 | 0.298 | −0.227 | −0.068 | 0.071 | ||
| 6a | −8.6751 | 0.112 | 0.257 | 0.013 | −0.100 | −0.145 | 1.7804 | 0.256 | −0.215 | −0.153 | 0.126 | 0.041 |
| 6b | −8.1608 | 0.043 | 0.242 | 0.017 | −0.091 | −0.199 | 1.9179 | 0.272 | −0.210 | −0.151 | 0.122 | 0.062 |
| 6c | −8.8008 | 0.106 | 0.246 | 0.012 | −0.094 | −0.140 | 1.5032 | 0.248 | −0.215 | −0.142 | 0.119 | 0.033 |
| 6d | −9.5564 | 0.148 | 0.271 | 0.005 | −0.108 | −0.123 | 0.5236 | −0.164 | 0.183 | 0.095 | −0.088 | −0.019 |
| 4a a | 5a a | 6a a | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diene | HOMO-LUMO | LUMO-HOMO | Diff. | HOMO-LUMO | LUMO-HOMO | Diff. | HOMO-LUMO | LUMO-HOMO | Diff. |
| 1 | 9.8155 | 11.8447 | 2.0292 | 9.3382 | 12.1299 | 2.7917 | 10.5855 | 11.5816 | 0.9961 |
| 2 | 9.5714 | 12.0417 | 2.4703 | 9.0941 | 12.3269 | 3.2328 | 10.3414 | 11.7786 | 1.4372 |
| 3 | 9.6512 | 11.6626 | 2.0114 | 9.1739 | 11.9478 | 2.7739 | 10.4212 | 11.3995 | 0.9783 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Flores-Conde, M.I.; Reyes, L.; Herrera, R.; Rios, H.; Vazquez, M.A.; Miranda, R.; Tamariz, J.; Delgado, F. Highly Regio- and Stereoselective Diels-Alder Cycloadditions via Two-Step and Multicomponent Reactions Promoted by Infrared Irradiation under Solvent-Free Conditions. Int. J. Mol. Sci. 2012, 13, 2590-2617. https://doi.org/10.3390/ijms13032590
Flores-Conde MI, Reyes L, Herrera R, Rios H, Vazquez MA, Miranda R, Tamariz J, Delgado F. Highly Regio- and Stereoselective Diels-Alder Cycloadditions via Two-Step and Multicomponent Reactions Promoted by Infrared Irradiation under Solvent-Free Conditions. International Journal of Molecular Sciences. 2012; 13(3):2590-2617. https://doi.org/10.3390/ijms13032590
Chicago/Turabian StyleFlores-Conde, Maria Ines, Leonor Reyes, Rafael Herrera, Hulme Rios, Miguel A. Vazquez, Rene Miranda, Joaquin Tamariz, and Francisco Delgado. 2012. "Highly Regio- and Stereoselective Diels-Alder Cycloadditions via Two-Step and Multicomponent Reactions Promoted by Infrared Irradiation under Solvent-Free Conditions" International Journal of Molecular Sciences 13, no. 3: 2590-2617. https://doi.org/10.3390/ijms13032590
APA StyleFlores-Conde, M. I., Reyes, L., Herrera, R., Rios, H., Vazquez, M. A., Miranda, R., Tamariz, J., & Delgado, F. (2012). Highly Regio- and Stereoselective Diels-Alder Cycloadditions via Two-Step and Multicomponent Reactions Promoted by Infrared Irradiation under Solvent-Free Conditions. International Journal of Molecular Sciences, 13(3), 2590-2617. https://doi.org/10.3390/ijms13032590