Synthesis and Characterization of Privileged Monodentate Phosphoramidite Ligands and Chiral Brønsted Acids Derived from D-Mannitol


 , and
, and
Abstract
:
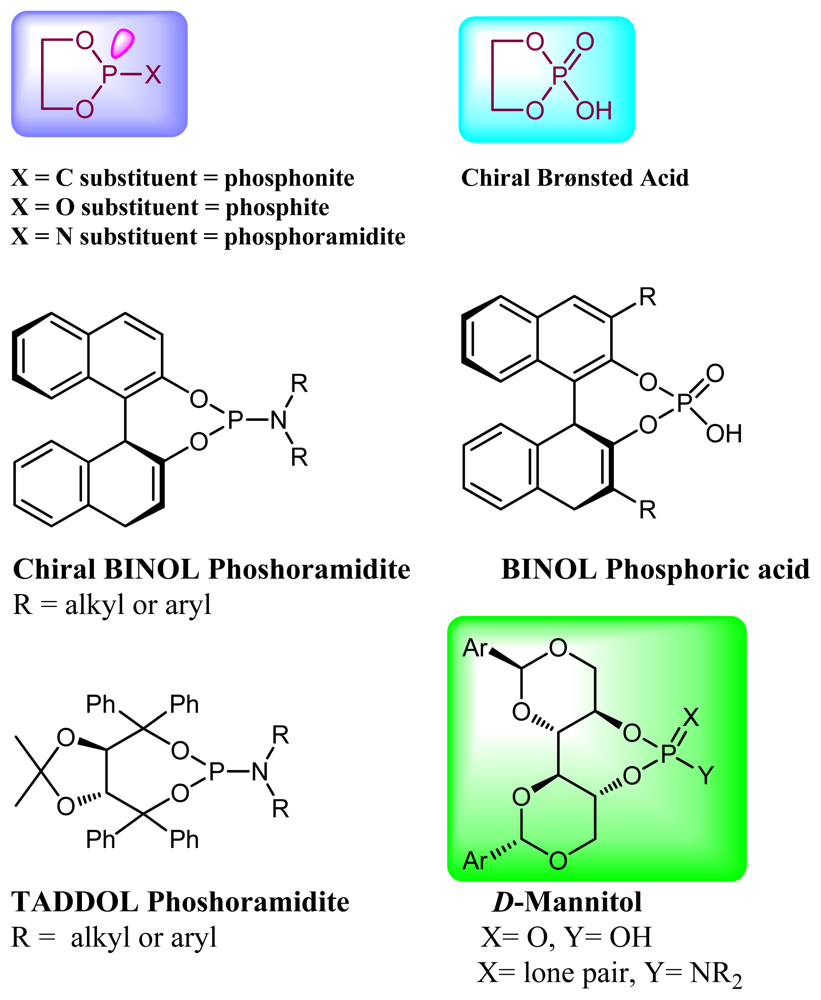
1. Introduction
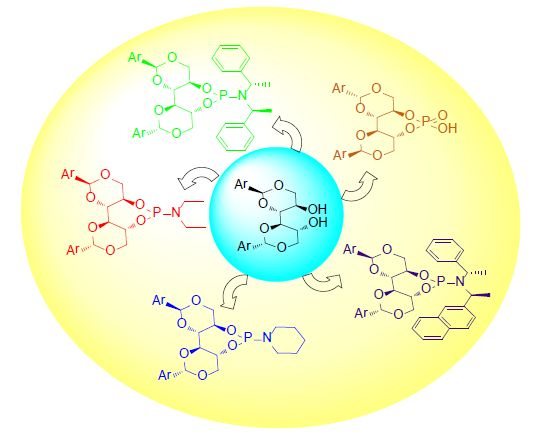
2. Results and Discussion
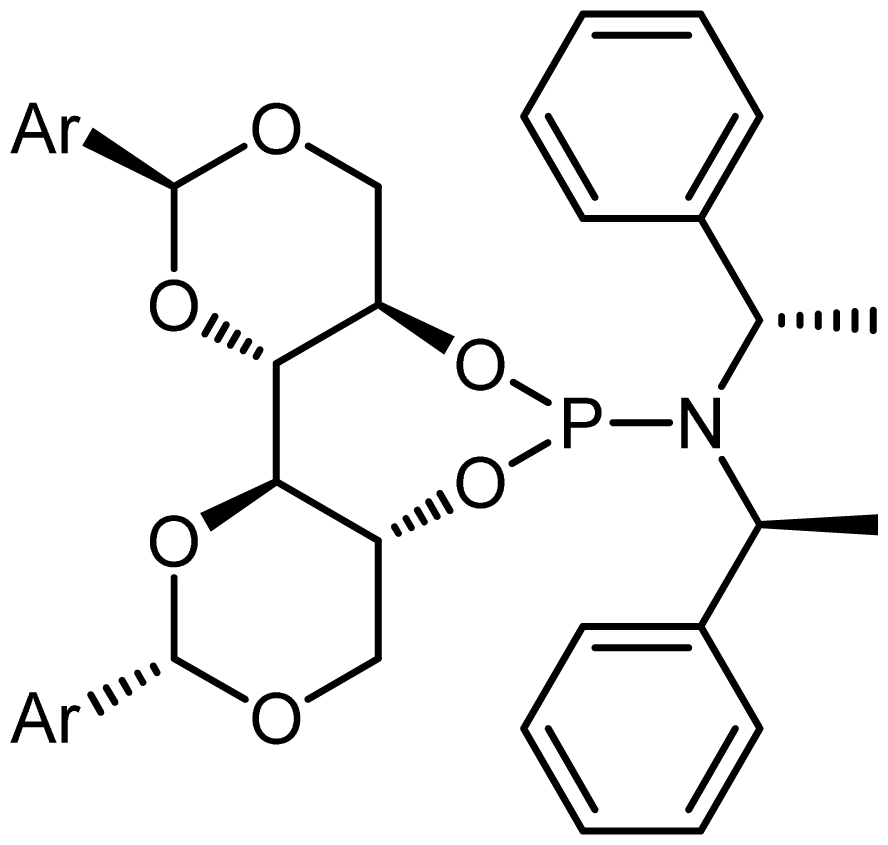
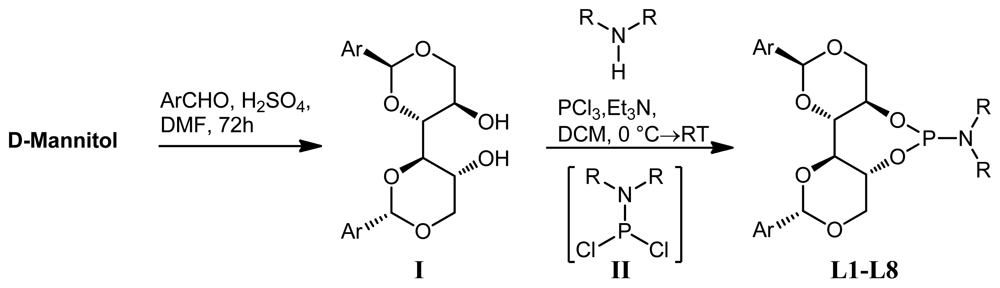
2.1. Synthesis of Phosphoramidite Ligands
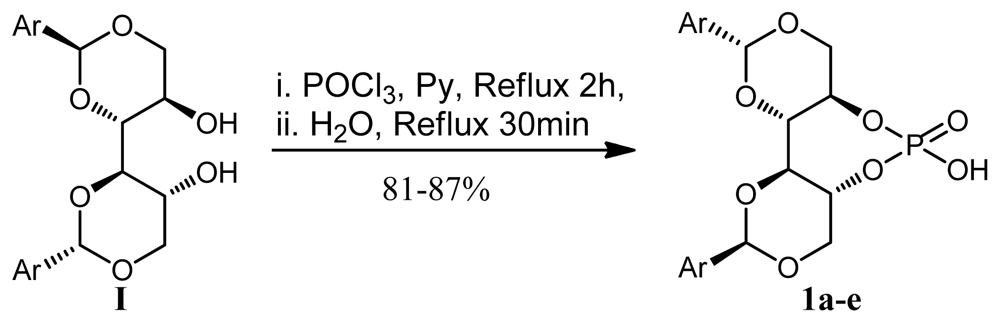
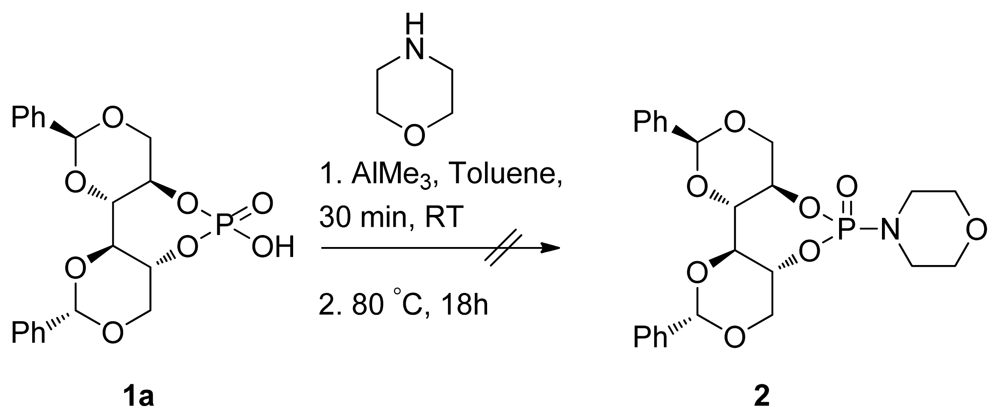
2.2. Synthesis of Chiral Brønsted Acids
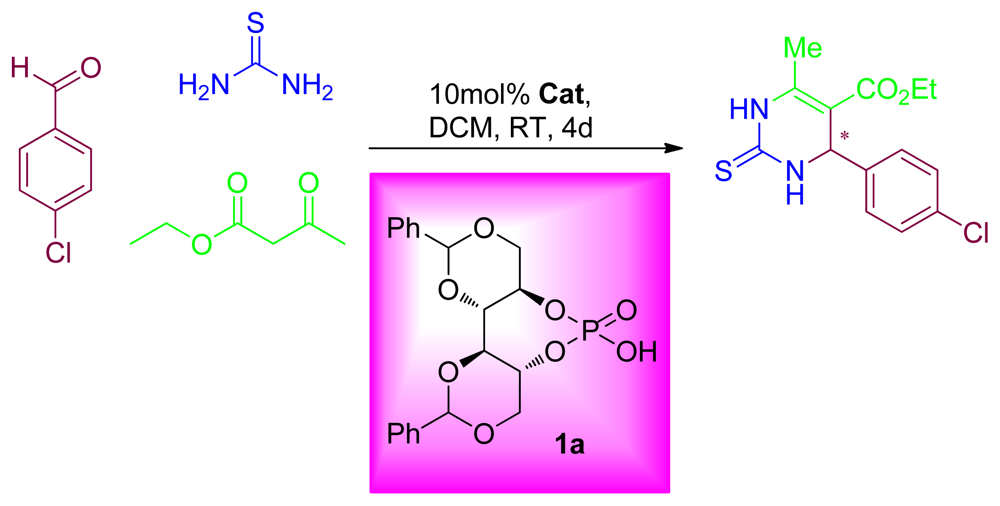
2.3. Applications
3. Experimental Section
3.1. General Procedure for the Synthesis of C2 Symmetric and Pseudo C2 Symmetric Secondary Amines (Procedure A) [35]
3.2. (R)-Bis((R)-1-Phenylethyl) Amine
3.3. (R)-1-(Naphthalen-2-yl)-N-((R)-1-Phenylethyl) Ethanamine
3.4. General Procedure for the Preparation of Phosphoramidites (Procedure B)
3.5. (4aR,7aR,11aS,11bS)-N,N-Diethyl-2,10-Diphenylhexahydrobis([1,3]Dioxino)[5,4-d:4′,5′- f][1,3,2]Dioxaphosphepin-6-amine (L1)
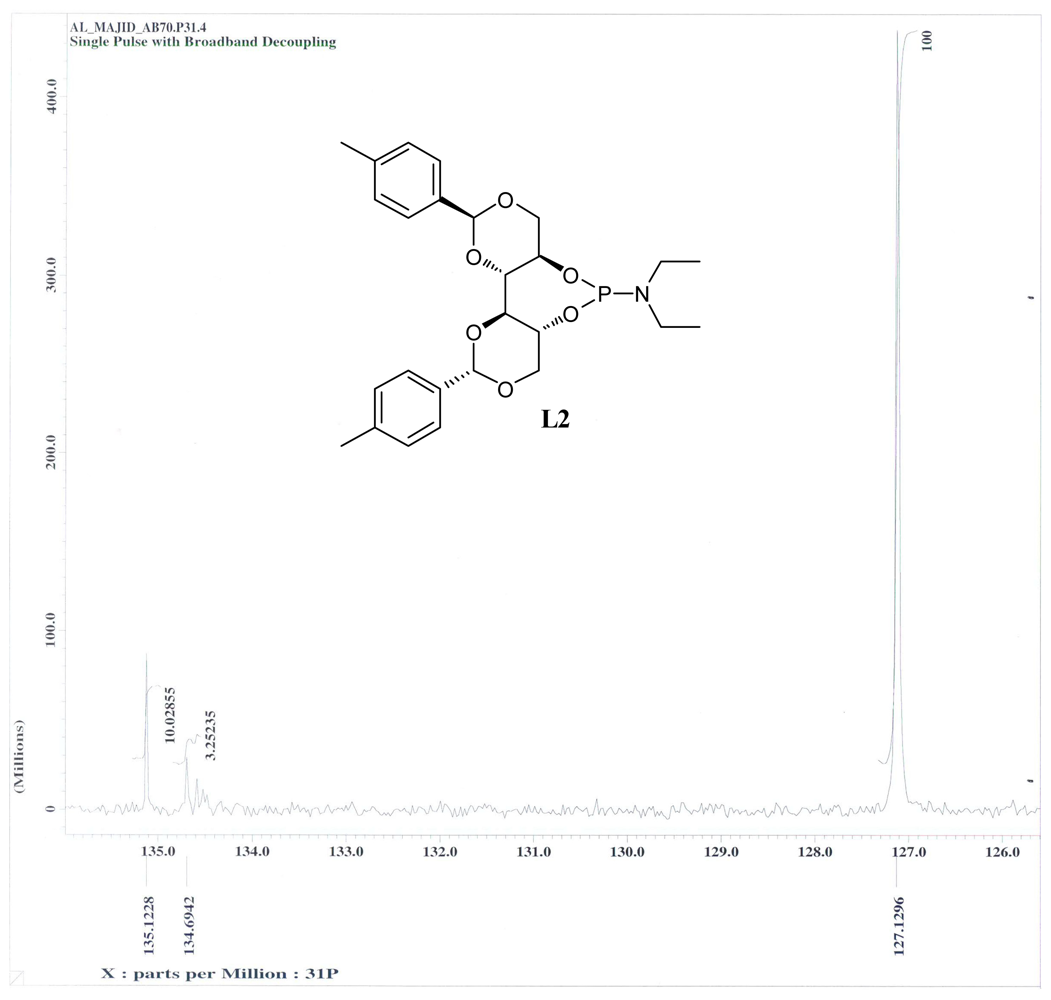
3.6. (4aR,7aR,11aS,11bS)-N,N-Diethyl-2,10-di-p-Tolylhexahydrobis([1,3]Dioxino)[5,4-d:4′,5′- f][1,3,2]Dioxaphosphepin-6-amine (L2)
3.7. 1-((4aR,7aR,11aS,11bS)-2,10-Diphenylhexahydrobis([1,3]dioxino)[5,4-d:4′,5′- f][1,3,2]dioxaphosphepin-6-yl)piperidine (L3)
3.8. 1-((4aR,7aR,11aS,11bS)-2,10-Di-p-Tolylhexahydrobis([1,3]dioxino)[5,4-d:4′,5′- f][1,3,2]dioxaphosphepin-6-yl)piperidine (L4)
3.9. (4aR,7aR,11aS,11bS)-2,10-Diphenyl-N,N-bis((S)-1-phenylethyl)hexahydrobis([1,3]dioxino) [5,4-d:4′,5′-f][1,3,2]dioxaphosphepin-6-amine (L5)
3.10. (4aR,7aR,11aS,11bS)-N,N-Bis((S)-1-Phenylethyl)-2,10-di-ptolylhexahydrobis([ 1,3]dioxino)[5,4-d:4′,5′-f][1,3,2]dioxaphosphepin-6-amine (L6)
3.11. (4aR,7aR,11aS,11bS)-N-((S)-1-(Naphthalen-2-yl)ethyl)-2,10-diphenyl-N-((S)-1- Phenylethyl)hexahydrobis([1,3]dioxino)[5,4-d:4′,5′-f][1,3,2]dioxaphosphepin-6-amine (L7)
3.12. (4aR,7aR,11aS,11bS)-N-((S)-1-(Naphthalen-2-yl)ethyl)-N-((S)-1-phenylethyl)-2,10-di-ptolylhexahydrobis([ 1,3]dioxino)[5,4-d:4′,5′-f][1,3,2]dioxaphosphepin-6-amine (L8)
3.13. General Procedure for the Preparation of Chiral Brønsted Acid (Procedure C) [20]
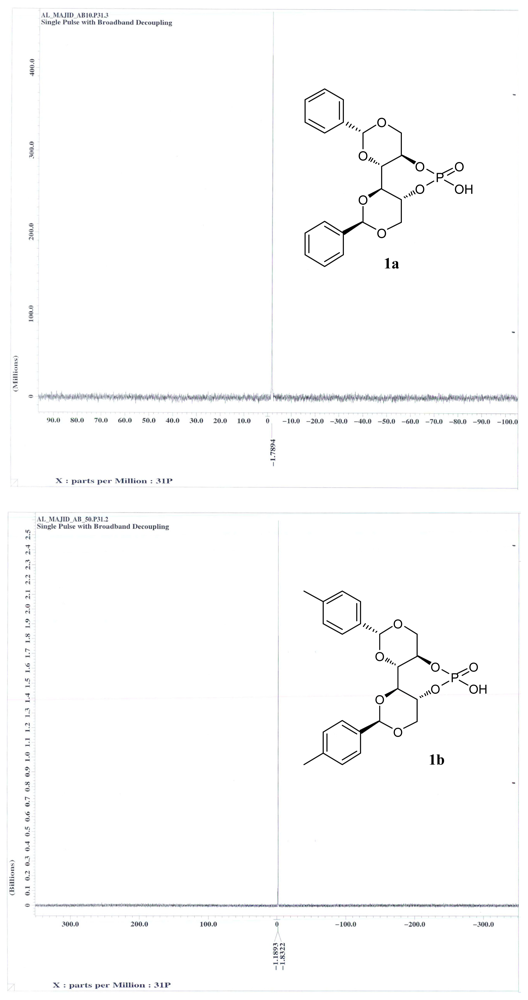
3.14. (4aR,7aR,11aS,11bS)-6-Hydroxy-2,10-diphenylhexahydrobis([1,3]dioxino)[5,4-d:4′,5′- f][1,3,2]Dioxaphosphepine 6-Oxide (1a)
3.15. (4aR,7aR,11aS,11bS)-6-Hydroxy-2,10-di-p-tolylhexahydrobis([1,3]dioxino)[5,4-d:4′,5′- f][1,3,2]Dioxaphosphepine 6-Oxide (1b)
4. Conclusions
Acknowledgments
References
- Noyori, R. Centenary Lecture. Chemical multiplication of chirality: Science and applications. Chem. Soc. Rev 1989, 18, 187–208. [Google Scholar]
- Jacobsen, E.N.; Pfaltz, A.; Yamamoto, H. Comprehensive Asymmetric Catalysis I–III; Springer: Berlin, Germany, 1999. [Google Scholar]
- Eberhardt, L.; Armspach, D.; Harrowfield, J.; Matt, D. BINOL-derived phosphoramidites in asymmetric hydrogenation: Can the presence of a functionality in the amino group influence the catalytic outcome? Chem. Soc. Rev 2008, 37, 839–864. [Google Scholar]
- Toselli, N.; Martin, D.; Achard, M.; Tenaglia, A.; Burgi, T.; Buono, G. Enantioselective cobalt-catalyzed [6+2] cycloadditions of cycloheptatriene with alkynes. Adv. Synth. Catal 2008, 350, 280–286. [Google Scholar]
- Kostas, I.D.; Vallianatou, K.A.; Holz, J.; Borner, A. A new easily accessible chiral phosphate-phosphoramidite ligand based on 2-anilinoethanol and R-BINOL moieties for Rh-catalyzed asymmetric olefin hydrogenation. Tetrahedron Lett 2008, 49, 331–334. [Google Scholar]
- Mršić, N.; Lefort, L.; Boogers, J.A.F.; Minnaard, A.J.; Feringa, B.L.; de Vries, J.G. Asymmetric hydrogenation of quinolines catalyzed by iridium complexes of monodentate BINOL-derived phosphoramidites. Adv. Synth. Catal 2008, 350, 1081–1089. [Google Scholar]
- Minnaard, A.J.; Feringa, B.L.; Lefort, L.; de Vries, J.G. Asymmetric hydrogenation using monodentate phosphoramidite ligands. Acc. Chem. Res 2007, 40, 1267–1277. [Google Scholar]
- Zhang, W.; Zhang, X. Highly enantioselective hydrogenation of α-dehydroamino esters and itaconates with triphosphorous bidentate ligands and the unprecedented solvent effect thereof. J. Org. Chem 2007, 72, 1020–1023. [Google Scholar]
- Shekar, S.; Trantow, B.; Leitner, A.; Hartwig, J.F. Sequential catalytic isomerization and allylic substitution. Conversion of racemic branched allylic carbonates to enantioenriched allylic substitution products. J. Am. Chem. Soc 2006, 128, 11770–11771. [Google Scholar]
- Zhao, B.; Wang, Z.; Ding, K. Practical by ligand design: A new class of monodentate phosphoramidite ligands for rhodium-catalyzed enantioselective hydrogenations. Adv. Synth. Catal 2006, 348, 1049–1057. [Google Scholar]
- Alexakis, A.; Albrow, V.; Biswas, K.; d’Augustin, M.; Prietob, O.; Woodward, S. Highly enantioselective copper(I)-phosphoramidite-catalysed additions of organoaluminium reagents to enones. Chem. Commun 2005, 2843–2845. [Google Scholar]
- van den Berg, M.; Minnaard, A.J.; Haak, R.M.; Leeman, M.; Schudde, E.P.; Meetsma, A.; Feringa, B.L.; de Vries, A.H.M.; Maljaars, C.E.P.; Willans, C.E.; et al. Monodentate phosphoramidites: A breakthrough in rhodium-catalysed asymmetric hydrogenation of olefins. Adv. Synth. Catal 2003, 345, 308–323. [Google Scholar]
- DeVries, A.H.M.; Meetsma, A.; Feringa, B.L. Enantioselective conjugate addition of dialkylzinc reagents to cyclic and acyclic enones catalyzed by chiral copper complexes of new phosphorus amidites. Angew. Chem. Int. Ed 1996, 35, 2375–2376. [Google Scholar]
- Mikhel, I.S.; Ruegger, H.; Butti, P.; Camponovo, F.; Huber, D.; Mezzetti, A. A chiral phosphoramidite beyond monodentate coordination: Secondary π-interactions turn a dangling aryl into a two-, four-, or six-electron donor in d6 and d8 complexes. Organometallics 2008, 27, 2937–2948. [Google Scholar]
- Sakaki, J.-I.; Schweizer, W.B.; Seebach, D. Catalytic enantioselective hydrosilylation of aromatic ketones using rhodium complexes of TADDOL-derived cyclic phosphonites and phosphites. Helv. Chim. Acta 1993, 76, 2654–2665. [Google Scholar]
- Alexakis, A.; Frutos, J.; Mangeney, P. Chiral phosphorus ligands for the asymmetric conjugate addition of organocopper reagents. Tetrahedron Asymmetry 1993, 4, 2427–2430. [Google Scholar]
- Alexakis, A.; Bäckvall, J.E.; Krause, N.; Pàmies, O.; Diéguez, M. Enantioselective copper-catalyzed conjugate addition and allylic substitution reactions. Chem. Rev 2008, 108, 2796–2823. [Google Scholar]
- Feringa, B.L. Phosphoramidites: Marvellous ligands in catalytic asymmetric conjugate addition. Acc. Chem. Res 2000, 33, 346–353. [Google Scholar]
- Minnaard, A.J.; Feringa, B.L.; Lefort, L.; de Vries, J.G. Asymmetric hydrogenation using monodentate phosphoramidite ligands. Acc. Chem. Res 2007, 40, 1267–1277. [Google Scholar]
- Teichert, J.F.; Feringa, B.L. Phosphoramidites: Privileged ligands in asymmetric catalysis. Angew. Chem. Int. Ed 2010, 49, 2486–2528. [Google Scholar]
- Peña, D.; Minnaard, A.J.; de Vries, J.G.; Feringa, B.L. Highly enantioselective rhodium-catalyzed hydrogenation of beta-dehydroamino acid derivatives using monodentate phosphoramidites. J. Am. Chem. Soc 2002, 124, 14552–14553. [Google Scholar]
- Burks, H.E.; Liu, S.; Morken, J.P. Development, mechanism, and scope of the palladium-catalyzed enantioselective allene diboration. J. Am. Chem. Soc 2007, 129, 8766–8773. [Google Scholar]
- Albicker, M.R.; Cramer, N. Enantioselective palladium-catalyzed direct arylations at ambient temperature: Access to indanes with quaternary stereocenters. Angew. Chem. Int. Ed 2009, 48, 9139–9142. [Google Scholar]
- Bayer, A.; Thewalt, U.; Rieger, B. Novel monodentate phosphoramidites by chiral pool synthesis starting from d-mannitol, and their PdII and RhI complexes. Eur. J. Inorg. Chem 2002, 1, 199–203. [Google Scholar]
- Maciá, B.; Fernández-Ibáñez, A.; Mršić, N.; Minnaard, A.M.; Feringa, B.L. Copper-catalyzed enantioselective conjugate addition of Grignard reagents to acyclic enones using monodentate phosphoramidite ligands. Tetrahedron Lett 2008, 49, 1877–1880. [Google Scholar]
- Pfretzschner, T.; Kleemann, L.; Janza, B.; Harms, K.; Schrader, T. On the role of PIII ligands in the conjugate addition of diorganozinc derivatives to enones. Chem. Eur. J 2004, 10, 6048–6057. [Google Scholar]
- Jensen, J.F.; Svendsen, B.Y.; la Cour, T.V.; Pedersen, H.L.; Johannsen, M. Highly enantioselective hydrosilylation of aromatic alkenes. J. Am. Chem. Soc 2002, 124, 4558–4559. [Google Scholar]
- Duursma, A.; Boiteau, J.-G.; Lefort, L.; Boogers, J.A.F.; de Vries, A.H.M.; de Vries, J.G.; Minnaard, A.J.; Feringa, B.L. Highly enantioselective conjugate additions of potassium organotrifluoroborates to enones by use of monodentate phosphoramidite ligands. J. Org. Chem 2004, 69, 8045–8052. [Google Scholar]
- Trost, B.M.; Silverman, S.M.; Stambuli, J.P. Palladium-catalyzed asymmetric [3+2] cycloaddition of trimethylenemethane with imines. J. Am. Chem. Soc 2007, 129, 12398–12399. [Google Scholar]
- Trost, B.M.; Cramer, N.; Silverman, S.M. Enantioselective construction of spirocyclic oxindolic cyclopentanes by palladium-catalyzed trimethylenemethane-[3+2]-cycloaddition. J. Am. Chem. Soc 2007, 129, 12396–12397. [Google Scholar]
- Trost, B.M.; Stambuli, J.P.; Silverman, S.M.; Schwörer, U. Palladium-catalyzed asymmetric [3+2] trimethylenemethane cycloaddition reactions. J. Am. Chem. Soc 2006, 128, 13328–13329. [Google Scholar]
- Faller, J.W.; Fontaine, P.P. Resolution and diels-alder catalysis with planar chiral arene-tethered ruthenium complexes. Organometallics 2005, 24, 4132–4138. [Google Scholar]
- Imbos, R.; Minnaard, A.J.; Feringa, B.L. Monodentate phosphoramidites: Versatile ligands in catalytic asymmetric intramolecular Heck reactions. Dalton Trans 2003, 10. [Google Scholar] [CrossRef]
- Baggett, N.; Stribblehill, P. Asymmetric reduction of ketones by using complexes of lithium tetrahydridoaluminate(III) with 1,4:3,6-dianhydro-d-mannitol and 1,3:4,6-di-O-benzylidene-d-mannitol. J. Chem. Soc. Perkin Trans 1997, 1, 1123–1126. [Google Scholar]
- Trost, B.M.; Silverman, S.M.; Stambuli, J.P. Development of an asymmetric trimethylenemethane cycloaddition reaction: Application in the enantioselective synthesis of highly substituted carbocycles. J. Am. Chem. Soc 2011, 133, 19483–19497. [Google Scholar]
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev 2007, 107, 5713–5743. [Google Scholar]
- Akiyama, T. Stronger Brønsted acids. Chem. Rev 2007, 107, 5744–5758. [Google Scholar]
- Yu, X.; Wang, W. Hydrogen-bond-mediated asymmetric catalysis. Chem. Asian J 2008, 3, 516–532. [Google Scholar]
- Terada, M. Binaphthol-derived phosphoric acid as a versatile catalyst for enantioselective carbon-carbon bond forming reactions. Chem. Commun 2008, 4097–4112. [Google Scholar]
- Vachal, P.; Jacobsen, E.N. Structure-based analysis and optimization of a highly enantioselective catalyst for the strecker reaction. J. Am. Chem. Soc 2002, 124, 10012–10014. [Google Scholar]
- Zhang, X.; Du, H.; Wang, Z.; Wu, Y.-D.; Ding, K. Experimental and theoretical studies on the hydrogen-bond-promoted enantioselective hetero-Diels-Alder reaction of Danishefsky’s diene with benzaldehyde. J. Org. Chem 2006, 71, 2862–2869. [Google Scholar]
- Gridnev, I.D.; Kouchi, M.; Sorimachi, K.; Terada, M. On the mechanism of stereoselection in direct Mannich reaction catalyzed by BINOL-derived phosphoric acids. Tetrahedron Lett 2007, 48, 497–500. [Google Scholar]
- Yamanaka, M.; Itoh, J.; Fuchibe, K.; Akiyama, T. Chiral Brønsted acid catalyzed enantioselective mannich-type reaction. J. Am. Chem. Soc 2007, 129, 6756–6764. [Google Scholar]
- Simón, L.; Goodman, J.M. Theoretical study of the mechanism of hantzsch ester hydrogenation of imines catalyzed by chiral BINOL-phosphoric acids. J. Am. Chem. Soc 2008, 130, 8741–8747. [Google Scholar]
- Anderson, C.D.; Dudding, T.; Gordillo, R.; Houk, K.N. Origin of enantioselection in hetero-Diels-Alder reactions catalyzed by naphthyl-TADDOL. Org. Lett 2008, 10, 2749–2752. [Google Scholar]
- Corey, E.J.; Lee, T. The formyl C–H…O hydrogen bond as a critical factor in enantioselective Lewis-acid catalyzed reactions of aldehydes. Chem. Commun 2001, 1321–1329. [Google Scholar]
- Castellano, R.K. Progress toward understanding the nature and function of C–H…O interactions. Curr. Org. Chem 2004, 8, 845–865. [Google Scholar]
- Washington, I.; Houk, K.N. CH…O Hydrogen bonding influences π-facial stereoselective epoxidations. Angew. Chem. Int. Ed 2001, 40, 4485–4488. [Google Scholar]
- Terada, M.; Soga, K.; Momiyama, N. Enantioselective activation of aldehydes by chiral phosphoric acid catalysts in an Aza-ene-type reaction between glyoxylate and enecarbamate. Angew. Chem. Int. Ed 2008, 47, 4122–4125. [Google Scholar]
- Kashikura, W.; Mori, K.; Akiyama, T. Chiral phosphoric acid catalyzed enantioselective synthesis of β-amino-α,α-difluoro carbonyl compounds. Org. Lett 2011, 13, 1860–1863. [Google Scholar]
- Li, J.; Subramaniam, K.; Smith, D.; Qiao, J.X.; Li, J.J.; Q-Cutrone, J.; Kadow, J.F.; Vite, G.D.; Chen, B-C. AlMe3-promoted formation of amides from acids and amines. Org. Lett. 2012, 14, 214–217. [Google Scholar]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Majid, A.M.A.; Barakat, A.; Mabkhot, Y.N.; Islam, M.S. Synthesis and Characterization of Privileged Monodentate Phosphoramidite Ligands and Chiral Brønsted Acids Derived from D-Mannitol. Int. J. Mol. Sci. 2012, 13, 2727-2743. https://doi.org/10.3390/ijms13032727
Al-Majid AMA, Barakat A, Mabkhot YN, Islam MS. Synthesis and Characterization of Privileged Monodentate Phosphoramidite Ligands and Chiral Brønsted Acids Derived from D-Mannitol. International Journal of Molecular Sciences. 2012; 13(3):2727-2743. https://doi.org/10.3390/ijms13032727
Chicago/Turabian StyleAl-Majid, Abdullah Mohammed A., Assem Barakat, Yahia Nasser Mabkhot, and Mohammad Shahidul Islam. 2012. "Synthesis and Characterization of Privileged Monodentate Phosphoramidite Ligands and Chiral Brønsted Acids Derived from D-Mannitol" International Journal of Molecular Sciences 13, no. 3: 2727-2743. https://doi.org/10.3390/ijms13032727