Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size

Abstract
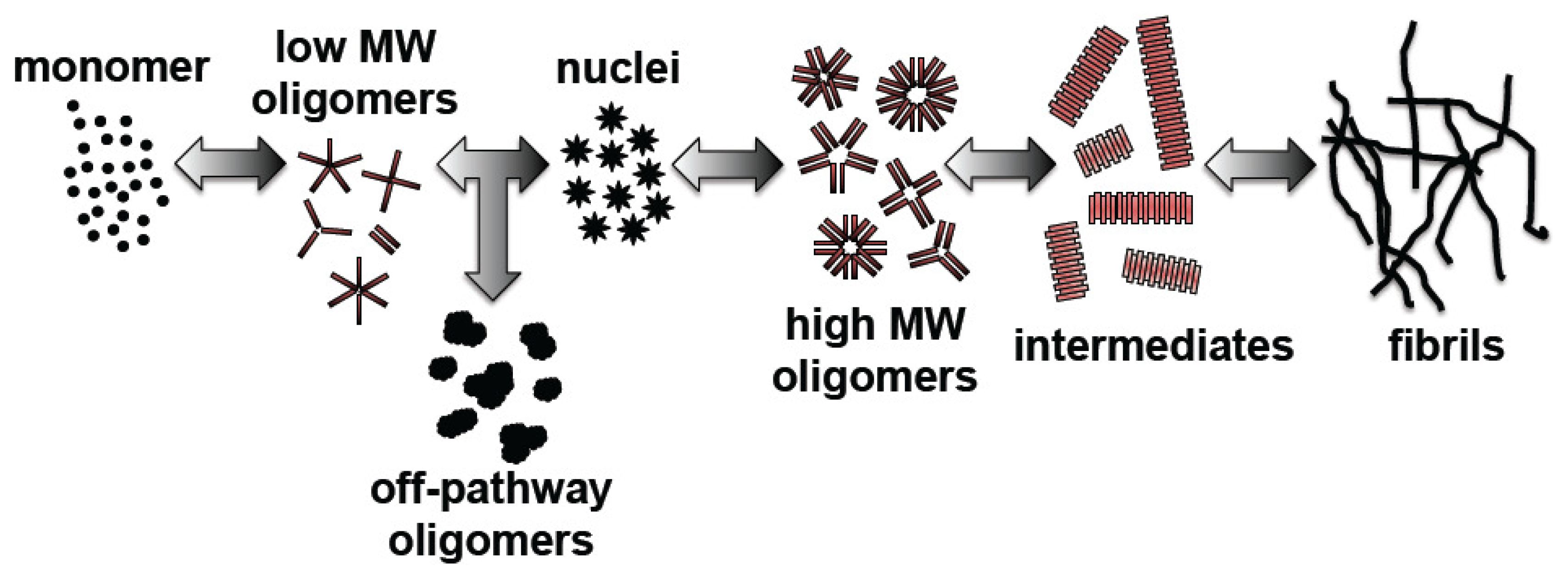
:1. Introduction
2. Electrophoretic Techniques for the Quantification of Aβ Oligomer Sizes
2.1. SDS- and Native-PAGE
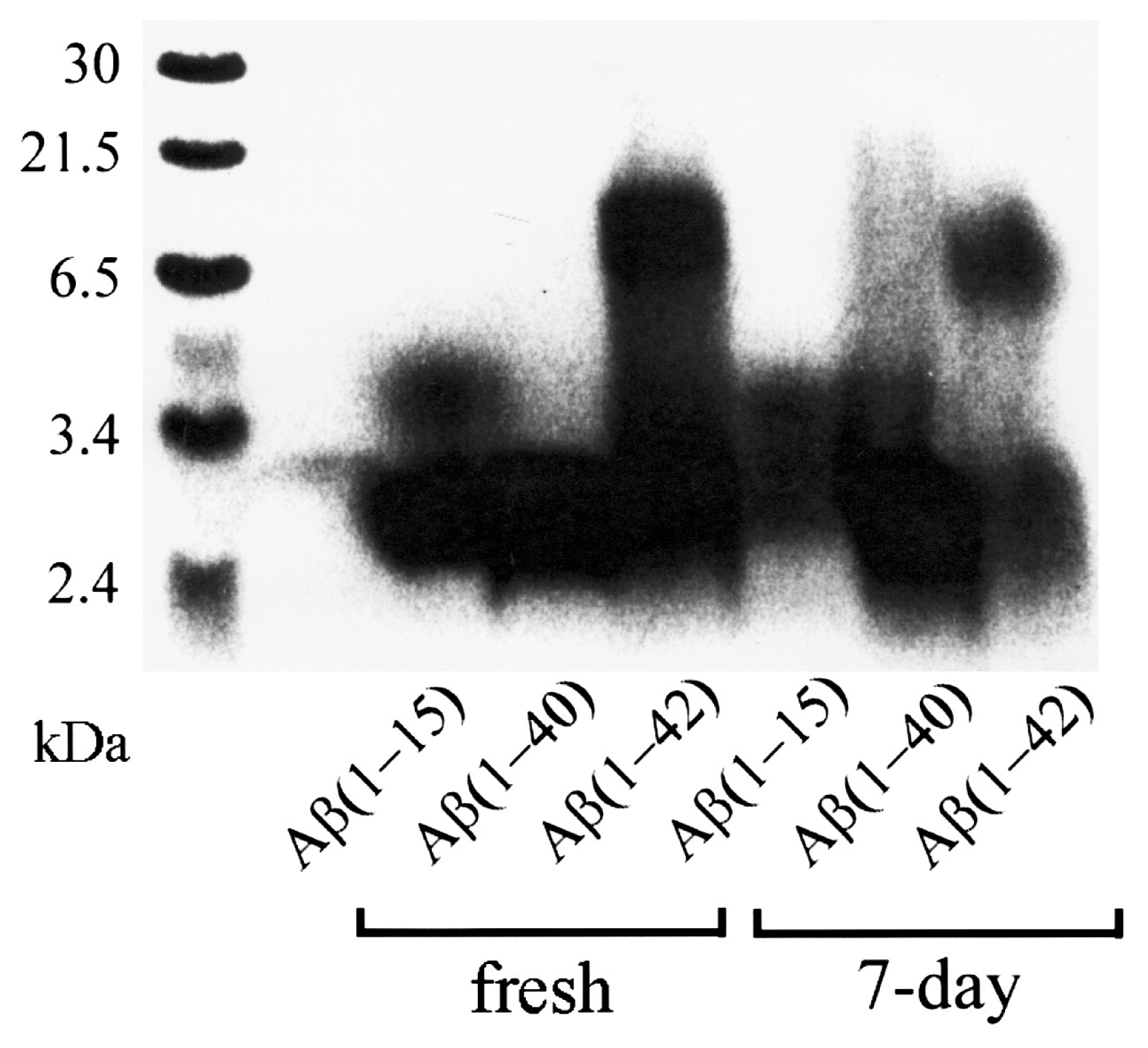
2.2. SDS-PAGE in Combination with Western Blotting
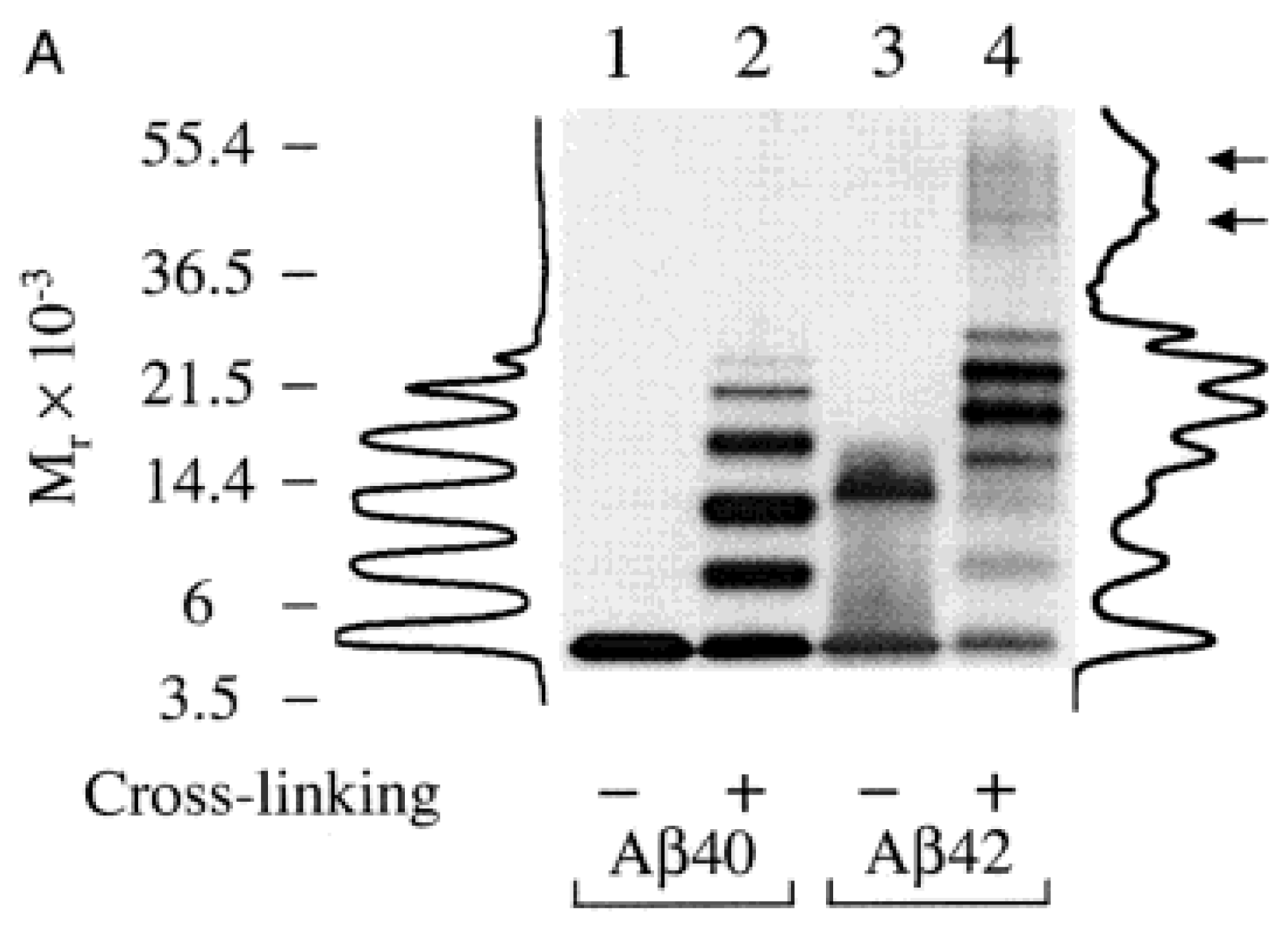
2.3. SDS-PAGE in Combination with Other Techniques
2.4. Summary of SDS-Based Methods
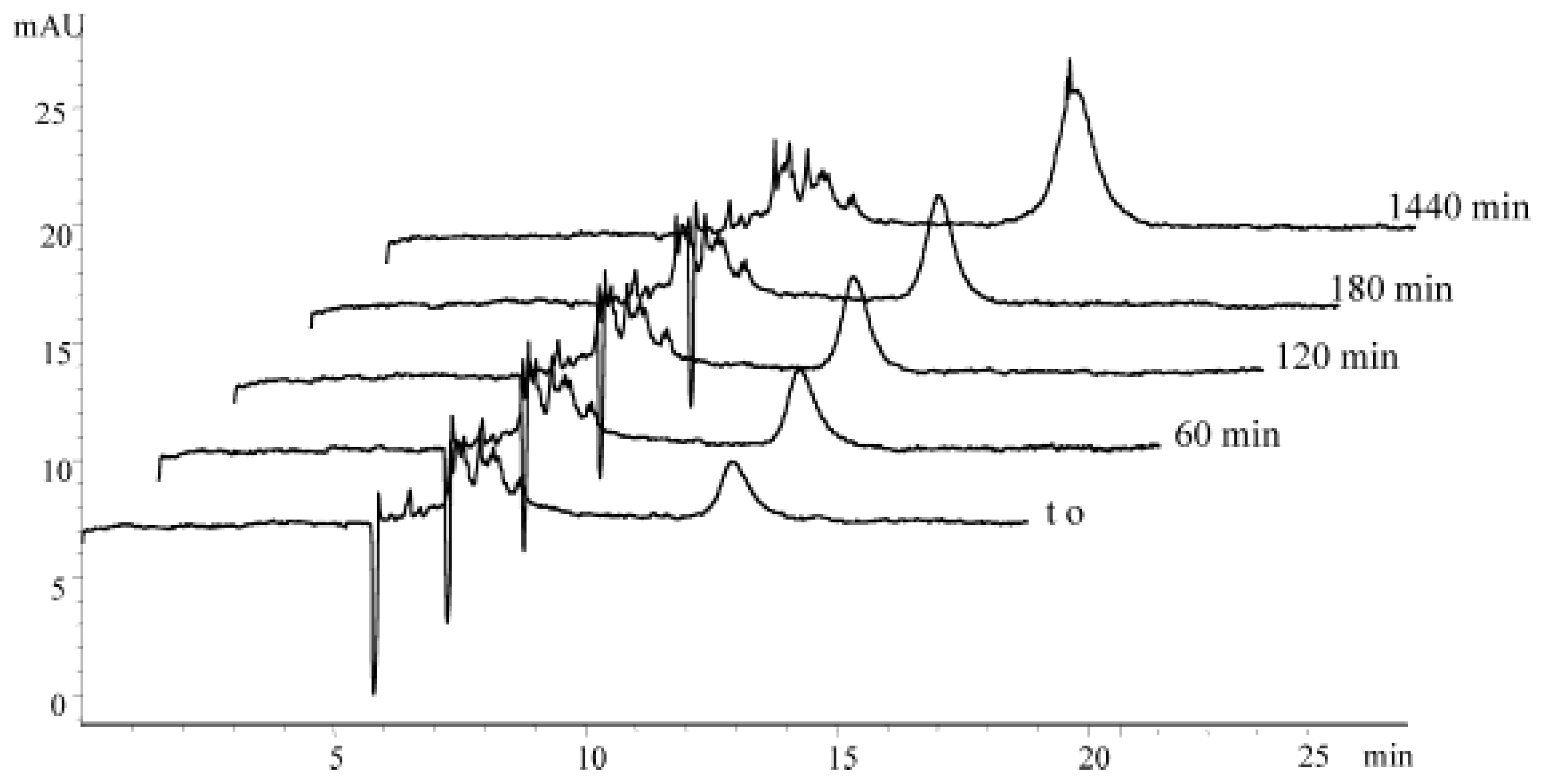
2.5. Capillary and Microfluidic Capillary Electrophoresis
3. Spectroscopic Techniques for the Quantification of Aβ Oligomer Sizes
3.1. Mass Spectrometry
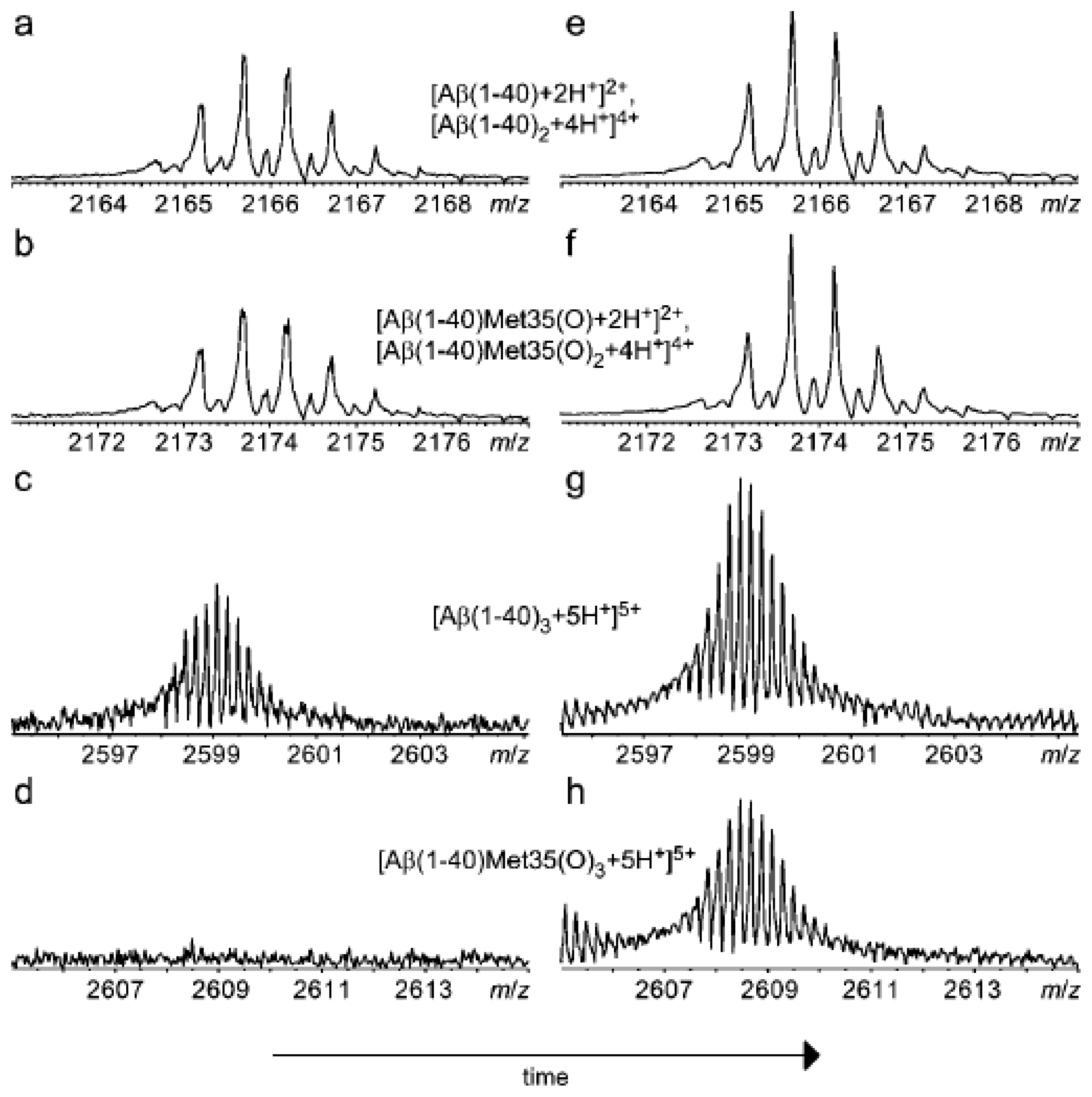
3.2 Matrix-Assisted Laser-Desorption Ionization (MALDI)-MS
3.3. Electrospray Ionization (ESI)-MS
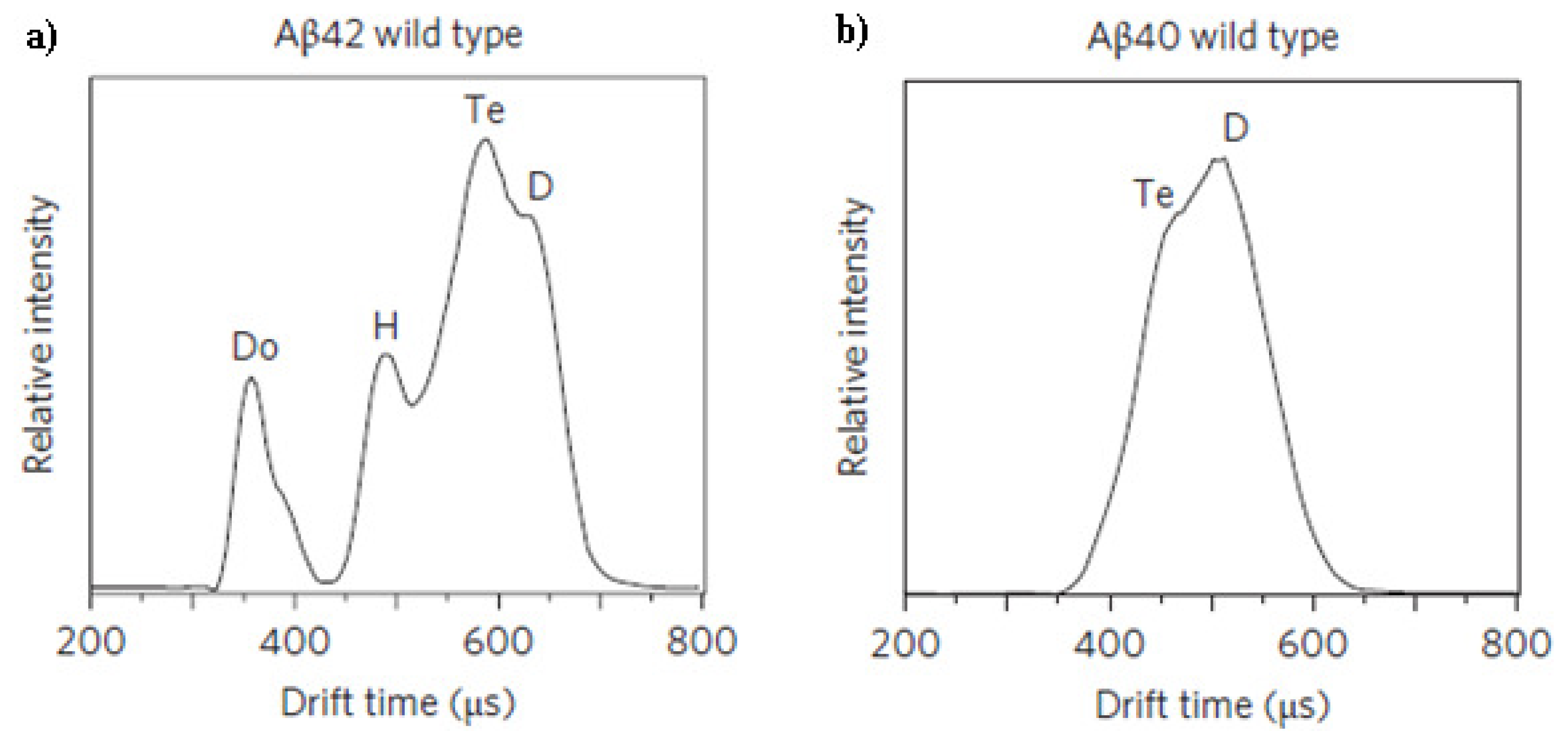
3.4. Ion Mobility (IM)-MS
3.5. Fluorescence Correlation Spectroscopy
3.6. Summary of Spectroscopic Methods
4. Additional Techniques Utilized for Aβ Aggregate Size Determinations
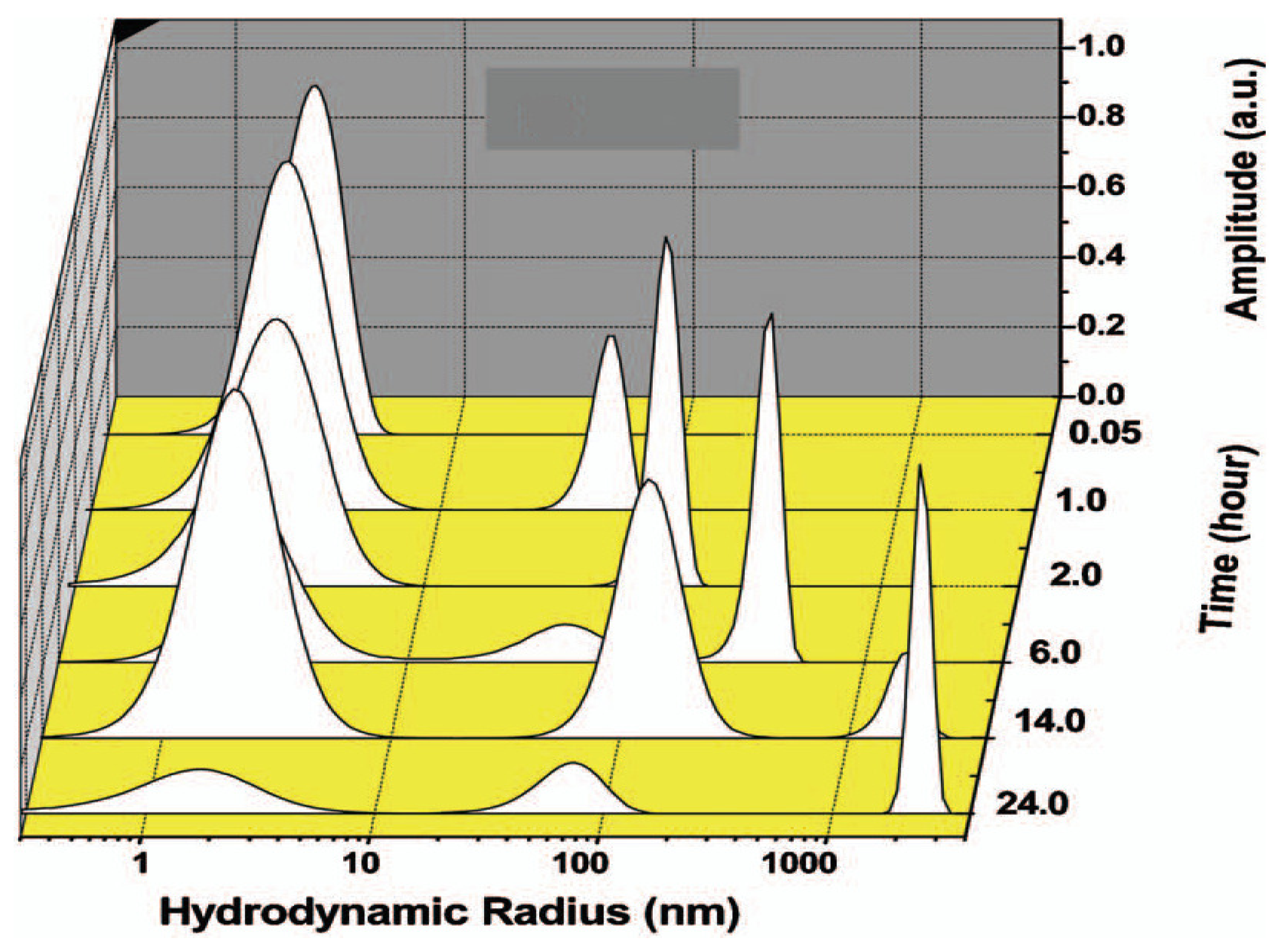
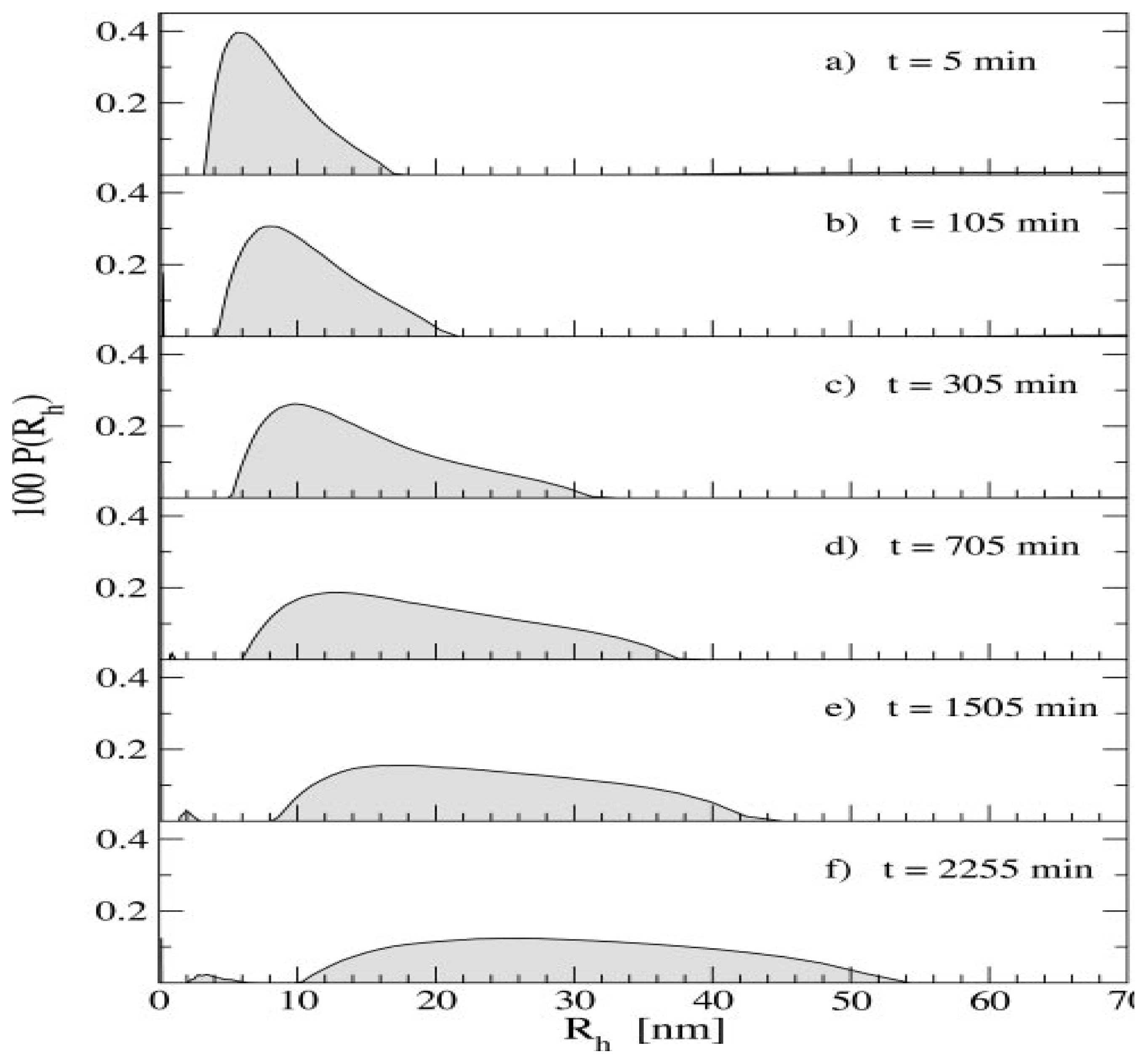
4.1. Light Scattering Techniques
4.2. Light Scattering in Combination with Other Techniques
4.3. Centrifugation
4.4. Size Exclusion Chromatography
4.5. Summary of Additional Aβ Aggregate Size Determination Techniques
5. Techniques Utilized for Aβ Oligomer Identification
5.1. Dot Blot
5.2. Enzyme-Linked Immunosorbent Assay
5.3. Summary of Aβ Oligomer Identification Techniques
6. Conclusions
Acknowledgements
References
- Robinson, J.L.; Geser, F.; Corrada, M.M.; Berlau, D.J.; Arnold, S.E.; Lee, V.M.; Kawas, C.H.; Trojanowski, J.Q. Neocortical and hippocampal amyloid-β and tau measures associate with dementia in the oldest-old. Brain 2011, 134, 3708–3715. [Google Scholar]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National institute on aging-alzheimer’s association guidelines for the neuropathologic assessment of alzheimer’s disease: A practical approach. Acta Neuropathol 2012, 123, 1–11. [Google Scholar]
- Kagan, B.L.; Jang, H.; Capone, R.; Arce, F.T.; Ramachandran, S.; Lal, R.; Nussinov, R. Antimicrobial properties of amyloid peptides. Mol. Pharm 2011, in press. [Google Scholar]
- Miranker, A.D. Unzipping the mysteries of amyloid fiber formation. Proc. Natl. Acad. Sci. USA 2004, 101, 4335–4336. [Google Scholar]
- Murphy, R.M. Peptide aggregation in neurodegenerative disease. Annu. Rev. Biomed. Eng 2002, 4, 155–174. [Google Scholar]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar]
- Gazit, E. Mechanisms of amyloid fibril self-assembly and inhibition. Model short peptides as a key research tool. FEBS J 2005, 272, 5971–5978. [Google Scholar]
- Xing, Y.; Higuchi, K. Amyloid fibril proteins. Mech. Ageing Dev 2002, 123, 1625–1636. [Google Scholar]
- Kisilevsky, R. Review: Amyloidogenesis-unquestioned answers and unanswered questions. J. Struct. Biol 2000, 130, 99–108. [Google Scholar]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener 2007, 2. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar]
- Alzheimer’s Association. 2011 alzheimer’s disease facts and figures. Alzheimers Dement. 2011, 7, 1–63.
- Wang, X.; Ding, H. Alzheimer’s disease: Epidemiology, genetics, and beyond. Neurosci. Bull 2008, 24, 105–109. [Google Scholar]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid Plaque core protein in alzheimer disease and down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar]
- Giuffrida, M.L.; Caraci, F.; de Bona, P.; Pappalardo, G.; Nicoletti, F.; Rizzarelli, E.; Copani, A. The monomer state of β-amyloid: Where the alzheimer’s disease protein meets physiology. Rev. Neurosci 2010, 21, 83–93. [Google Scholar]
- Teplow, D.B. Structural and kinetic features of amyloid β-protein fibrillogenesis. Int. J. Exp. Clin. Invest 1998, 5, 121–142. [Google Scholar]
- Morris, A.M.; Watzky, M.A.; Finke, R.G. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. biochim. biophys Acta Proteins Proteomics 2009, 1794, 375–397. [Google Scholar]
- Kodali, R.; Wetzel, R. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol 2007, 17, 48–57. [Google Scholar]
- Walsh, D.M.; Selkoe, D.J. Aβ oligomers—A decade of discovery. J. Neurochem 2007, 101, 1172–1184. [Google Scholar]
- Sabate, R.; Estelrich, J. Evidence of the existence of micelles in the fibrillogenesis of β-amyloid peptide. J. Phys. Chem. B 2005, 109, 11027–11032. [Google Scholar]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar]
- Catalano, S.M.; Dodson, E.C.; Henze, D.A.; Joyce, J.G.; Krafft, G.A.; Kinney, G.G. The role of Amyloid-β Derived Diffusible Ligands (ADDLs) in alzheimer’s disease. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2006, 6, 597–608. [Google Scholar]
- Westlind-Danielsson, A.; Arnerup, G. Spontaneous in vitro formation of supramolecular β-amyloid structures, “βamy Balls”, by β-amyloid 1-40 peptide. Biochemistry 2001, 40, 14736–14743. [Google Scholar]
- Matsumura, S.; Shinoda, K.; Yamada, M.; Yokojima, S.; Inoue, M.; Ohnishi, T.; Shimada, T.; Kikuchi, K.; Masui, D.; Hashimoto, S.; et al. Two distinct amyloid β-protein (Aβ) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology, and toxicity analyses. J. Biol. Chem 2011, 286, 11555–11562. [Google Scholar]
- Walsh, D.; Lomakin, A.; Benedek, G.; Condron, M.; Teplow, D. Amyloid β-protein fibrillogenesis: Detection of a protofibrillar intermediate. J. Biol. Chem 1997, 272, 22364–22372. [Google Scholar]
- Ward, R.; Jennings, K.; Howlett, D. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of β-amyloid peptide. Biochem. J 2000, 348. [Google Scholar]
- Thirumalai, D.; Klimov, D.; Dima, R. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr. Opin. Struct. Biol 2003, 13, 146–159. [Google Scholar]
- Roher, A.E.; Baudry, J.; Chaney, M.O.; Kuo, Y.-M.; Stine, W.B.; Emmerling, M.R. Oligomerization and Fibril Assembly of the Amyloid-β Protein. Biochim. Biophys. Acta Mol. Basis Dis 2000, 1502, 31–43. [Google Scholar]
- Morgan, C.; Colombres, M.; Nunez, M.T.; Inestrosa, N.C. Structure and function of amyloid in alzheimer’s disease. Prog. Neurobiol 2004, 74, 323–349. [Google Scholar]
- Barghorn, S.; Nimmrich, V.; Striebinger, A.; Krantz, C.; Keller, P.; Janson, B.; Bahr, M.; Schmidt, M.; Bitner, R.S.; Harlan, J.; et al. Globular amyloid β-peptide1-42 oligomer—A homogeneous and stable neuropathological protein in alzheimer’s disease. J. Neurochem 2005, 95, 834–847. [Google Scholar]
- Kawooya, J.; Emmons, T.; Gonzalez-DeWhitt, P.; Camp, M.; D’Andrea, S. Electrophoretic mobility of alzheimer’s amyloid-β peptides in urea-sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal. Biochem 2003, 323, 103–113. [Google Scholar]
- Gursky, O.; Aleshkov, S. Temperature-dependent β-sheet formation in β-amyloid aβ1-40 peptide in water: Uncoupling β-structure folding from aggregation. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzymol 2000, 1476, 93–102. [Google Scholar]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu Rev. Neurosci 2003, 26, 267–298. [Google Scholar]
- Glabe, C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 2006, 27, 570–575. [Google Scholar]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid β-protein assembly and alzheimer’s disease. J. Biol. Chem 2009, 284, 4749–4753. [Google Scholar]
- Gonzalez-Velasquez, F.J.; Kotarek, J.A.; Moss, M.A. Soluble aggregates of the amyloid-β protein selectively stimulate permeability in human brain microvascular endothelial monolayers. J. Neurochem 2008, 107, 466–477. [Google Scholar]
- Hartley, D.M.; Walsh, D.M.; Ye, C.P.; Diehl, T.; Vasquez, S.; Vassilev, P.M.; Teplow, D.B.; Selkoe, D.J. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci 1999, 19, 8876–8884. [Google Scholar]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Rowan, M.J.; Selkoe, D.J. Amyloid-β oligomers: Their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans 2002, 30, 552–557. [Google Scholar]
- Westerman, M.; Cooper-Blacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.; Carlson, G.; Younkin, S.; Ashe, K. The relationship between aβ and memory in the tg2576 mouse model of alzheimer’s disease. J. Neurosci 2002, 22, 1858–1867. [Google Scholar]
- Lue, L.F.; Kuo, Y.M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid β peptide concentration as a predictor of synaptic change in alzheimer’s disease. Am. J. Pathol 1999, 155, 853–862. [Google Scholar]
- Scheff, S.; Price, D.; Schmitt, F.; Mufson, E. Hippocampal synaptic loss in early alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar]
- Yang, T.; Hsu, C.; Kuo, Y. Cell-derived soluble oligomers of human amyloid-β peptides disturb cellular homeostasis and induce apoptosis in primary hippocampal neurons. J. Neural Transm 2009, 116, 1561–1569. [Google Scholar]
- Bitan, G.; Fradinger, E.A.; Spring, S.M.; Teplow, D.B. Neurotoxic protein oligomers—What you see is not always what you get. Amyloid 2005, 12, 88–95. [Google Scholar]
- Ying, Z.; Xin, W.; Jin-Sheng, H.; Fu-Xiang, B.; Wei-Min, S.; Xin-Xian, D.; Xiao-Bo, W.; Yi-Qin, L.; Xian-Xian, Z.; Hong-Gang, H.; et al. Preparation and characterization of a monoclonal antibody with high affinity for soluble Aβ oligomers. Hybridoma 2009, 28, 349–354. [Google Scholar]
- Satoh, Y.; Hirakura, Y.; Kirino, Y. Beta-amyloid peptides inhibit acetylcholine release from cholinergic presynaptic nerve endings isolated from an electric ray. Neurosci. Lett 2001, 302, 97–100. [Google Scholar]
- Sureshbabu, N.; Kirubagaran, R.; Jayakumar, R. Surfactant-induced conformational transition of amyloid β-peptide. Eur. Biophys. J 2009, 38, 355–367. [Google Scholar]
- Iurascu, M.; Cozma, C.; Tomczyk, N.; Rontree, J.; Desor, M.; Drescher, M.; Przybylski, M. Structural characterization of β-amyloid oligomer-aggregates by ion mobility mass spectrometry and electron spin resonance spectroscopy. Anal. Bioanal. Chem 2009, 395, 2509–2519. [Google Scholar]
- Klug, G.; Losic, D.; Small, D. Beta-amyloid protein oligomers induced by metal ions and acid pH are distinct from those generated by slow spontaneous ageing at neutral pH. Eur. J. Biochem 2003, 270, 4282–4293. [Google Scholar]
- Walsh, D.; Tseng, B.; Rydel, R.; Podlisny, M.; Selkoe, D. The oligomerization of amyloid β-protein begins intracellularly in cells derived from human brain. Biochemistry 2000, 39, 10831–10839. [Google Scholar]
- Stine, W.B., Jr; Dahlgren, K.N.; Krafft, G.A.; LaDu, M.J. In Vitro Characterization of Conditions for Amyloid-β Peptide Oligomerization and Fibrillogenesi. J. Biol. Chem. 2003, 278, 11612–11622. [Google Scholar]
- Ryan, D.; Narrow, W.; Federoff, H.; Bowers, W. An Improved Method for Generating Consistent Soluble Amyloid-β Oligomer Preparations for in Vitro Neurotoxicity Studies. J.Neurosci. Method 2010, 190, 171–179. [Google Scholar]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B., Jr; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and Fibrillar Species of Amyloid-β Peptides Differentially Affect Neuronal Viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar]
- Moore, B.; Rangachari, V.; Tay, W.; Milkovic, N.; Rosenberry, T. Biophysical Analyses of Synthetic Amyloid-β(1-42) Aggregates before and After Covalent Cross-Linking. Implications for Deducing the Structure of Endogenous Amyloid-β Oligomers. Biochemistry 2009, 48, 11796–11806. [Google Scholar]
- Walsh, D.; Hartley, D.; Condron, M.; Selkoe, D.; Teplow, D. In Vitro studies of Amyloid β-Protein Fibril Assembly and Toxicity Provide Clues to the Aetiology of Flemish Variant (Ala692 → Gly) Alzheimer’s Disease. Biochem. J 2001, 355, 869–877. [Google Scholar]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L., Jr; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid β protein (Aβ) in alzheimer’s disease brain. biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 Or Aβ42(43). J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar]
- Wong, H.E.; Qi, W.; Choi, H.; Fernandez, E.J.; Kwon, I. A safe, blood-brain barrier permeable triphenylmethane dye inhibits amyloid-β neurotoxicity by generating nontoxic aggregates. ACS Chem. Neurosci 2011, 2, 645–657. [Google Scholar]
- Hu, Y.; Su, B.H.; Kim, C.S.; Hernandez, M.; Rostagno, A.; Ghiso, J.; Kim, J.R. A strategy for designing a peptide probe for detection of β-amyloid oligomers. ChemBioChem 2010, 11, 2409–2418. [Google Scholar]
- Lambert, M.P.; Velasco, P.T.; Chang, L.; Viola, K.L.; Fernandez, S.; Lacor, P.N.; Khuon, D.; Gong, Y.; Bigio, E.; Shaw, P.; et al. Monoclonal antibodies that target pathological assemblies of Aβ. J. Neurochem 2007, 100, 23–25. [Google Scholar]
- Wu, J.W.; Breydo, L.; Isas, J.M.; Lee, J.; Kuznetsov, Y.G.; Langen, R.; Glabe, C. Fibrillar oligomers nucleate the oligomerization of monomeric amyloid β but do not seed fibril formation. J. Biol. Chem 2010, 285, 6071–6079. [Google Scholar]
- Zagorski, M.G.; Barrow, C.J. NMR studies of amyloid β-peptides: Proton assignments, secondary structure, and mechanism of an α-helix–α-sheet conversion for a homologous, 28-residue, N-terminal fragment. Biochemistry 1992, 31, 5621–5631. [Google Scholar]
- Bitan, G.; Teplow, D. Rapid photochemical cross-linking—A new tool for studies of metastable, amyloidogenic protein assemblies. Acc. Chem. Res 2004, 37, 357–364. [Google Scholar]
- Fancy, D.S.; Kodadek, T. Chemistry for the analysis of protein-protein interactions: Rapid and efficient cross-linking triggered by long wavelength light. Proc. Natl. Acad. Sci. USA 1999, 96, 6020–6024. [Google Scholar]
- Gerardi, R.D.; Barnett, N.W.; Lewis, S.W. Analytical Applications of Tris(2,2′-Bipyridyl) Ruthenium(III) as a Chemiluminescent Reagent. Anal. Chim. Acta 1999, 378, 1–43. [Google Scholar]
- Bitan, G.; Kirkitadze, M.; Lomakin, A.; Vollers, S.; Benedek, G.; Teplow, D. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar]
- Podlisny, M.; Walsh, D.; Selkoe, D. Oligomerization of endogeneous and synthetic amyloid β-protein at nanomolar levels in cell culture and stabilization of monomer by congo red. Biochemistry 1998, 37, 3602–3611. [Google Scholar]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol 2006, 572, 477–492. [Google Scholar]
- Zuberovic, A.; Hanrieder, J.; Wetterhall, M. Proteome Profiling of human cerebrospinal fluid: Exploring the potential of capillary electrophoresis with surface modified capillaries for analysis of complex biological samples. Eur. J. Mass Spectrom 2008, 14, 249–260. [Google Scholar]
- Lee, Y.-.l.; Maus, R.; Smith, B.; Winefordner, J. Laser-induced fluorescence detection of a single molecule in a capillary. Anal. Chem. 1994, 66, 4142–4149. [Google Scholar]
- Skeidsvoll, J.; Ueland, P. Analysis of double-stranded dna by capillary electrophoresis with laser-induced fluorescence detection using the monomeric dye SYBR green I. Anal. Biochem 1995, 231, 359–365. [Google Scholar]
- Verpillot, R.; Otto, M.; Taverna, M. Simultaneous analysis by capillary electrophoresis of five amyloid peptides as potential biomarkers of alzheimer’s disease. A J. Chromatogr 2008, 1214, 157–164. [Google Scholar]
- Sabella, S.; Quaglia, M.; Lanni, C.; Racchi, M.; Govoni, S.; Caccialanza, G.; Calligaro, A.; Bellotti, V.; Lorenzi, E. Capillary electrophoresis studies on the aggregation process of β-amyloid 1-42 and 1-40 peptides. Electrophoresis 2004, 25, 3186–3194. [Google Scholar]
- Picou, R.; Kheterpal, I.; Wellman, A.; Minnamreddy, M.; Ku, G.; Gilman, S.D. Analysis of Aβ(1-40) and Aβ(1-42) monomer and fibrils by capillary electrophoresis. J. Chromatogr. B 2011, 879, 627–632. [Google Scholar]
- Kato, M.; Kinoshita, H.; Toyo’oka, T. Analytical method for β-amyloid fibrils using ce-laser induced fluorescence and its application to screening for inhibitors of β-amyloid protein aggregation. Anal. Chem 2007, 79, 4887–4891. [Google Scholar]
- Jakeway, S.C.; de Mello, A.J.; Russell, E.L.; Fresenius, J. Miniaturized total analysis systems for biological analysis. Anal. Chem 2000, 366, 525–539. [Google Scholar]
- Chovan, T.; Guttman, A. Microfabricated devices in biotechnology and biochemical processing. Trends Biotechnol 2002, 20, 116–122. [Google Scholar]
- Mohamadi, M.R.; Svobodova, Z.; Verpillot, R.; Esselmann, H.; Wiltfang, J.; Otto, M.; Taverna, M.; Bilkova, Z.; Viovy, J. Microchip electrophoresis profiling of aβ peptides in the cerebrospinal fluid of patients with alzheimer’s disease. Anal. Chem 2010, 82, 7611–7617. [Google Scholar]
- Steiner, W.E.; Klopsch, S.J.; English, W.A.; Clowers, B.H.; Hill, H.H. Detection of a chemical warfare agent simulant in various aerosol matrixes by ion mobility time-of-flight mass spectrometry. Anal. Chem 2005, 77, 4792–4799. [Google Scholar]
- Huertas, M.L.; Marty, A.M.; Fontan, J.; Alet, I.; Duffa, G. Measurement of mobility and mass of atmospheric ions. J. Aerosol Sci 1971, 2, 145–150. [Google Scholar]
- Borsdorf, H.; Rudolph, M. Gas-phase ion mobility studies of constitutional isomeric hydrocarbons using different ionization techniques. Int. J. Mass Spectrom 2001, 208, 67–72. [Google Scholar]
- Hill, C.A.; Thomas, C.L.P. Programmable gate delayed ion mobility spectrometry-mass spectrometry: A study with low concentrations of dipropylene-glycol-monomethyl-ether in air. Analyst 2005, 130, 1155–1161. [Google Scholar]
- Sielemann, S.; Baumbach, J.I.; Schmidt, H.; Pilzecker, P. Quantitative analysis of benzene, toluene, and m-Xylene with the use of a UV-ion mobility spectrometer. Field Anal. Chem. Technol 2000, 4, 157–169. [Google Scholar]
- Valentine, S.J.; Anderson, J.G.; Ellington, A.D.; Clemmer, D.E. Disulfide-intact and -reduced lysozyme in the gas phase: Conformations and pathways of folding and unfolding. J. Phys. Chem 1997, 101, 3891–3900. [Google Scholar]
- Ells, B.; Barnett, D.A.; Froese, K.; Purves, R.W.; Hrudey, S.; Guevremont, R. Detection of chlorinated and brominated byproducts of drinking water disinfection using electrospray ionization-high-field asymmetric waveform ion mobility spectrometry-mass spectrometry. Anal. Chem 1999, 71, 4747–4752. [Google Scholar]
- Matz, L.M.; Hill, H.H.J. Evaluation of opiate separation by high-resolution electrospray ionization-ion mobility spectrometry/mass spectrometry. Anal. Chem 2001, 73, 1664–1669. [Google Scholar]
- Smith, D.P.; Giles, K.; Bateman, R.H.; Radford, S.E.; Ashcroft, A.E. Monitoring copopulated conformational states fduring protein folding events using electrospray ionization-ion mobility spectrometry-mass spectrometry. J. Am. Soc. Mass Spectrom 2007, 18, 2180–2190. [Google Scholar]
- Gillig, K.J.; Ruotolo, B.; Stone, E.G.; Russell, D.H.; Fuhrer, K.; Gonin, M.; Schultz, A.J. Coupling high-pressure MALDI with ion mobility/orthogonal time-of-flight mass spectrometry. Anal. Chem 2000, 72, 3965–3971. [Google Scholar]
- Woods, A.S.; Koomen, J.M.; Ruotolo, B.T.; Gillig, K.J.; Russel, D.H.; Fuhrer, K.; Gonin, M.; Egan, T.F.; Schultz, J.A. A study of peptide-peptide interactions using MALDI ion mobility o-TOF and ESI mass spectrometry. J. Am. Soc. Mass Spectrom 2002, 13, 166–169. [Google Scholar]
- Steiner, W.E.; Clowers, B.H.; English, W.A.; Hill, H.H.J. Atmospheric pressure matrix-assisted laser desorption/ionization with analysis by ion mobility time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom 2004, 18, 882–888. [Google Scholar]
- McLean, J.A.; Ruotolo, B.T.; Gillig, K.J.; Russell, D.H. Ion mobility-mass spectrometry: A new paradigm for proteomics. Int. J. Mass Spectrom 2005, 240, 301–315. [Google Scholar]
- von Helden, G.; Gotts, N.G.; Bowers, M.T. Experimental evidence for the formation of fullerenes by collisional heating of carbon rings in the gas phase. Nature 1993, 363, 60–63. [Google Scholar]
- von Helden, G.; Kemper, P.R.; Gotts, N.G.; Bowers, M.T. Isomers of small carbon cluster anions: Linear chains with up to 20 atoms. Science 1993, 259, 1300–1302. [Google Scholar]
- Jarrold, M.F.; Constant, V.A. Silicon cluster ions: Evidence for a structural transition. Phys. Rev. Lett 1991, 67, 2994–2997. [Google Scholar]
- Kanu, A.B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H.H. Ion mobility-mass spectrometry. J. Mass Spectrom 2008, 43, 1–22. [Google Scholar]
- Maji, S.; Ogorzalek Loo, R.; Inayathullah, M.; Spring, S.; Vollers, S.; Condron, M.; Bitan, G.; Loo, J.; Teplow, D. Amino acid position-specific contributions to amyloid β-protein oligomerization. J. Biol. Chem 2009, 284, 23580–23591. [Google Scholar]
- Oe, T.; Ackermann, B.L.; Inoue, K.; Berna, M.J.; Garner, C.O.; Gelfanova, V.; Dean, R.A.; Siemers, E.R.; Holtzman, D.M.; Farlow, M.R.; et al. Quantitative analysis of amyloid ? Peptides in cerebrospinal fluid of alzheimer’s disease patients by immunoaffinity purification and stable isotope dilution liquid chromatography/negative electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom 2006, 20, 3723–3735. [Google Scholar]
- Ashcroft, A. Mass spectrometry and and amyloid problem-how far can we go in the gas phase? J. Am. Soc. Mass Spectrom 2010, 21, 1087–1096. [Google Scholar]
- Palmblad, M.; Westlind-Danielsson, A.; Bergquist, J. Oxidation of methionine 35 attenuates formation of amyloid β-peptide 1-40 oligomers. J. Biol. Chem 2002, 277, 19506–19510. [Google Scholar]
- Clemmer, D.E.; Hudgins, R.R.; Jarrold, M.F. Naked protein conformations: Cytochrome c in the gas phase. J. Am. Chem. Soc 1995, 117, 10141–10142. [Google Scholar]
- Clemmer, D.E.; Jarrold, M.F. Ion mobility measurements and their applications to clusters and biomolecules. J. Mass Spectrom 1997, 32, 577–592. [Google Scholar]
- Henderson, S.C.; Valentine, S.J.; Counterman, A.E.; Clemmer, D.E. ESI/ion trap/ion mobility/ time-of-flight mass spectrometry for rapid and sensitive analysis of biomolecular mixtures. Anal. Chem 1999, 71, 291–301. [Google Scholar]
- Murray, M.; Bernstein, S.; Nyugen, V.; Condron, M.; Teplow, D.; Bowers, M. Amyloid β protein: Aβ40 inhibits Aβ42 oligomerization. J. Am. Chem. Soc 2009, 131, 6316–6317. [Google Scholar]
- Bernstein, S.; Wyttenbach, T.; Baumketner, A.; Shea, J.; Bitan, G.; Teplow, D.; Bowers, M. Amyloid β-protein: Monomer structure and early aggregation states of Aβ42 and its Pro19 alloform. J. Am. Chem. Soc 2005, 127, 2075–2084. [Google Scholar]
- Bernstein, S.; Dupuis, N.; Lazo, N.; Wyttenbach, T.; Condron, M.; Bitan, G.; Teplow, D.; Shea, J.; Ruotolo, B.; Robinson, C.; et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of alzheimer’s disease. Nat. Chem 2009, 1, 326–331. [Google Scholar]
- Cizas, P.; Budvytyte, R.; Morkuniene, R.; Moldovan, R.; Broccio, M.; Loesche, M.; Niaura, G.; Valincius, G.; Borutaite, V. Size-dependent neurotoxicity of β-amyloid oligomers. Arch. Biochem. Biophys 2010, 496, 84–92. [Google Scholar]
- Tjernberg, L.O.; Pramanik, A.; Bjorling, S.; Thyberg, P.; Thyberg, J.; Nordstedt, C.; Berndt, K.D.; Terenius, L.; Rigler, R. Amyloid β-peptide polymerization studied using fluorescence correlation spectroscopy. Chem. Biol 1999, 6, 53–62. [Google Scholar]
- Garai, K.; Sahoo, B.; Sengupta, P.; Maiti, S. Quasihomogeneous nucleation of amyloid β yields numerical bounds for the critical radius, the surface tension, and the free energy barrier for nucleus formation. J. Chem. Phys 2008, 128, 045102:1–045102:7. [Google Scholar]
- Eigen, M.; Rigler, R. Sorting single molecules: Application to diagnostics and evolutionary biotechnology. Proc. Natl. Acad. Sci. USA 1994, 91, 5740–5747. [Google Scholar]
- Garai, K.; Sengupta, P.; Sahoo, B.; Maiti, S. Selective destabilization of soluble amyloid β oligomers by divalent metal ions. Biochem. Biophys. Res. Commun 2006, 345, 210–215. [Google Scholar]
- Funke, S.A.; Birkmann, E.; Henke, F.; Goertz, P.; Lange-Asschenfeldt, C.; Riesner, D.; Willbold, D. Single particle detection of Aβ aggregates associated with alzheimer’s disease. Biochem. Biophys. Res. Commun 2007, 364, 902–907. [Google Scholar]
- Godderz, L.J.; Peak, M.M.; Rodgers, K.K. Analysis of biological macromolecular assemblies using static light scattering methods. Curr. Org. Chem 2005, 9, 899–908. [Google Scholar]
- Villari, V.; Micali, N. Light scattering as spectroscopic tool for the study of disperse systems useful in pharmaceutical sciences. J. Pharm. Sci 2008, 97, 1703–1730. [Google Scholar]
- Alexander, M.; Dalgleish, D. Dynamic light scattering techniques and their applications in food science. Food Biophys 2006, 1, 2–13. [Google Scholar]
- Lomakin, A.; Teplow, D.B.; Kirschner, D.A.; Benedek, G.B. Kinetic theory of fibrillogenesis of amyloid β-protein. Proc. Natl. Acad. Sci. USA 1997, 94, 7942–7947. [Google Scholar]
- Lomakin, A.; Chung, D.S.; Benedek, G.B.; Kirschner, D.A.; Teplow, D.B. On the nucleation and growth of amyloid β-protein fibrils: Detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. USA 1996, 93, 1125–1129. [Google Scholar]
- Thunecke, M.; Lobbia, A.; Kosciessa, U.; Dyrks, T.; Oakley, A.E.; Turner, J.; Saenger, W.; Georgalis, Y. Aggregation of Aβ alzheimer’s disease-related peptide studied by dynamic light scattering. J. Pept. Res 1998, 52, 509–517. [Google Scholar]
- Carrotta, R.; Manno, M.; Bulone, D.; Martorana, V.; San Biagio, P.L. Protofibril Formation of Amyloid β-Protein at Low pH via a Non-Cooperative Elongation Mechanism. J. Biol. Chem 2005, 280, 30001–30008. [Google Scholar]
- Murphy, R.M.; Pallitto, M.M. Probing the kinetics of β-amyloid self-association. J. Struct. Biol 2000, 130, 109–122. [Google Scholar]
- Rambaldi, D.C.; Zattoni, A.; Reschiglian, P.; Colombo, R.; de Lorenzi, E. In vitro amyloid Aβ(1-42) peptide aggregation monitoring by asymmetrical flow field-flow fractionation with multi-angle light scattering detection. Anal. Bioanal. Chem 2009, 394, 2145–2149. [Google Scholar]
- Nichols, M.; Moss, M.; Rosenberry, T. Growth of β-amyloid(1-40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry 2002, 41, 6115–6127. [Google Scholar]
- Walsh, D.; Hartley, D.; Kusumoto, Y.; Fezoui, Y.; Condron, M.; Lomakin, A.; Benedek, G.; Selkoe, D.; Teplow, D. Amyloid β-protein fibrillogenesis: Structure and biological activity of protofibrillar intermediates. J. Biol. Chem 1999, 274, 25945–25952. [Google Scholar]
- Yohannes, G.; Jussila, M.; Hartonen, K.; Riekkola, M. Asymmetrical flow field-flow fractionation technique for separation and characterization of biopolymers and bioparticles. J. Chromatogr. A 2011, 1218, 4104–4116. [Google Scholar]
- Roda, B.; Zattoni, A.; Reschiglian, P.; Moon, M.H.; Mirasoli, M.; Michelini, E.; Roda, A. Field-flow fractionation in bioanalysis: A review of recent trends. Anal. Chim. Acta 2009, 635, 132–143. [Google Scholar]
- Mok, Y.; Howlett, G.J. Sedimentation velocity analysis of amyloid oligomers and fibrils. Methods Enzymol 2006, 413, 199–217. [Google Scholar]
- Huang, T.H.J.; Yang, D.; Plaskos, N.P.; Go, S.; Yip, C.M.; Fraser, P.E.; Chakrabartty, A. Structural studies of soluble oligomers of the alzheimer β-amyloid peptide. J. Mol. Biol 2000, 297, 73–87. [Google Scholar]
- Nagel-Steger, L.; Demeler, B.; Meyer-Zaika, W.; Hochdoerffer, K.; Schrader, T.; Willbold, D. Modulation of aggregate size- and shape-distributions of the amyloid-β peptide by a designed β-sheet breaker. Eur. Biophys. J 2010, 39, 415–422. [Google Scholar]
- Paivio, A.; Jarvet, J.; Graslund, A.; Lannfelt, L.; Westlind-Danielsson, A. Unique physicochemical profile of β-amyloid peptide variant Aβ1-40E22G protofibrils: Conceivable neuropathogen in arctic mutant carriers. J. Mol. Biol 2004, 339, 145–159. [Google Scholar]
- Wiberg, H.; Ek, P. Separation and characterization of aggregated species of amyloid-β peptides. Anal. Bioanal. Chem 2010, 397, 2357–2366. [Google Scholar]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β1-42 oligomers to fibrils. Nat. Struct. Mol. Biol 2010, 17, 561–567. [Google Scholar]
- Englund, H.; Sehlin, D.; Johansson, A.; Nilsson, L.N.G.; Gellerfors, P.; Paulie, S.; Lannfelt, L.; Pettersson, F.E. Sensitive ELISA detection of amyloid-β protofibrils in biological samples. J. Neurochem 2007, 103, 334–345. [Google Scholar]
- Zheng, X.; Wang, L.; Zhang, L.; Hong, Y.; Huang, L.; Sha, Y. Separation and analysis of the soluble trimer of Aβ1-40 and its effects on the rise in intracellular calcium. Chin. Sci. Bull 2006, 51, 830–838. [Google Scholar]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem 2007, 282, 10311–10324. [Google Scholar]
- Gonzales, A.M.; Orlando, R.A. A sensitive Aβ oligomerization assay for identification of small molecule inhibitors. Open Biotechnol. J 2009, 3, 108–116. [Google Scholar]
- Kamali-Moghaddam, M.; Pettersson, F.E.; Wu, D.; Englund, H.; Darmanis, S.; Lord, A.; Tavoosidana, G.; Sehlin, D.; Gustafsdottir, S.; Nilsson, L.N.G.; et al. Sensitive detection of Aβ protofibrils by proximity ligation—Relevance for alzheimer’s disease. BMC Neurosci 2010, 11. [Google Scholar] [CrossRef]
- Stenh, C.; Englund, H.; Lord, A.; Johansson, A.; Almeida, C.G.; Gellerfors, P.; Greengard, P.; Gouras, G.K.; Lannfelt, L.; Nilsson, L.N.G. Amyloid-β oligomers are inefficiently measured by enzyme-linked immunosorbent assay. Ann. Neurol 2005, 58, 147–150. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Recognition Motif | Monoclonal/Polyclonal | References |
|---|---|---|---|
| 6E10 | Aβ1-17 | Monoclonal | [50–53] |
| Ab9 | Aβ1-16 | Monoclonal | [54] |
| 6C6 | Aβ1-16 | Monoclonal | [50] |
| 4G8 | Aβ17-24 | Monoclonal | [50] |
| 2G3 | Aβ31-40 | Monoclonal | [55] |
| BA-27 | Aβ1-40, C-terminal | Monoclonal | [56] |
| BC-05 | Aβ1-42, C-terminal | Monoclonal | [56] |
| A8 | amyloid oligomers | Monoclonal | [45] |
| A11 | amyloid oligomers | Monoclonal | [10,57,58] |
| NU-4 | amyloid oligomers | Monoclonal | [59] |
| OC | amyloid fibrils | Polycolonal | [60] |
| Technique | Advantages | Disadvantages | Aggregate Sizes Detected | References |
|---|---|---|---|---|
| SDS-PAGE |
|
| 4.5–20 kDa, >83 kDa | [45,46] |
| Native PAGE |
|
| 8–20 kDa, high molecular weight | [48,49] |
| Western Blotting |
|
| 4–16 kDa, 16.5–25 kDa, 30–97 kDa (with SDS-PAGE) | [45,50–53,55] |
| Capillary and Microfluidic Capillary Electrophoresis |
|
| 4–50 kDa, >50 kDa, fibrils | [72–74] |
| Mass Spectrometry |
|
| 4–24 kDa, ~48 kDa, fibrils, | [48,95,98, 102–104] |
| Fluorescence Correlation Spectroscopy |
|
| ~10 nm–1 μm (small oligomers– aggregates) | [24,105,107] |
| Light Scattering |
|
| >10 kDa (MALS) 1 nm–1 μm (DLS) | [112,116,117] |
| Centrifugation |
|
| 4–17 kDa, >250 kDa, ~270 kDa–3.8 MDa | [26,126] |
| Size Exclusion Chromatography |
|
| 4–20 kDa, 24 kDa, >100 kDa | [129–131] |
| Enzyme-Linked Immunosorbent Assay |
|
| No size determination | [130,133] |
| Dot Blot |
|
| No size determination | [57,132] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pryor, N.E.; Moss, M.A.; Hestekin, C.N. Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size. Int. J. Mol. Sci. 2012, 13, 3038-3072. https://doi.org/10.3390/ijms13033038
Pryor NE, Moss MA, Hestekin CN. Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size. International Journal of Molecular Sciences. 2012; 13(3):3038-3072. https://doi.org/10.3390/ijms13033038
Chicago/Turabian StylePryor, N. Elizabeth, Melissa A. Moss, and Christa N. Hestekin. 2012. "Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size" International Journal of Molecular Sciences 13, no. 3: 3038-3072. https://doi.org/10.3390/ijms13033038
APA StylePryor, N. E., Moss, M. A., & Hestekin, C. N. (2012). Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size. International Journal of Molecular Sciences, 13(3), 3038-3072. https://doi.org/10.3390/ijms13033038