Prediction of Genomic Islands in Three Bacterial Pathogens of Pneumonia

Abstract
:1. Introduction
2. Results
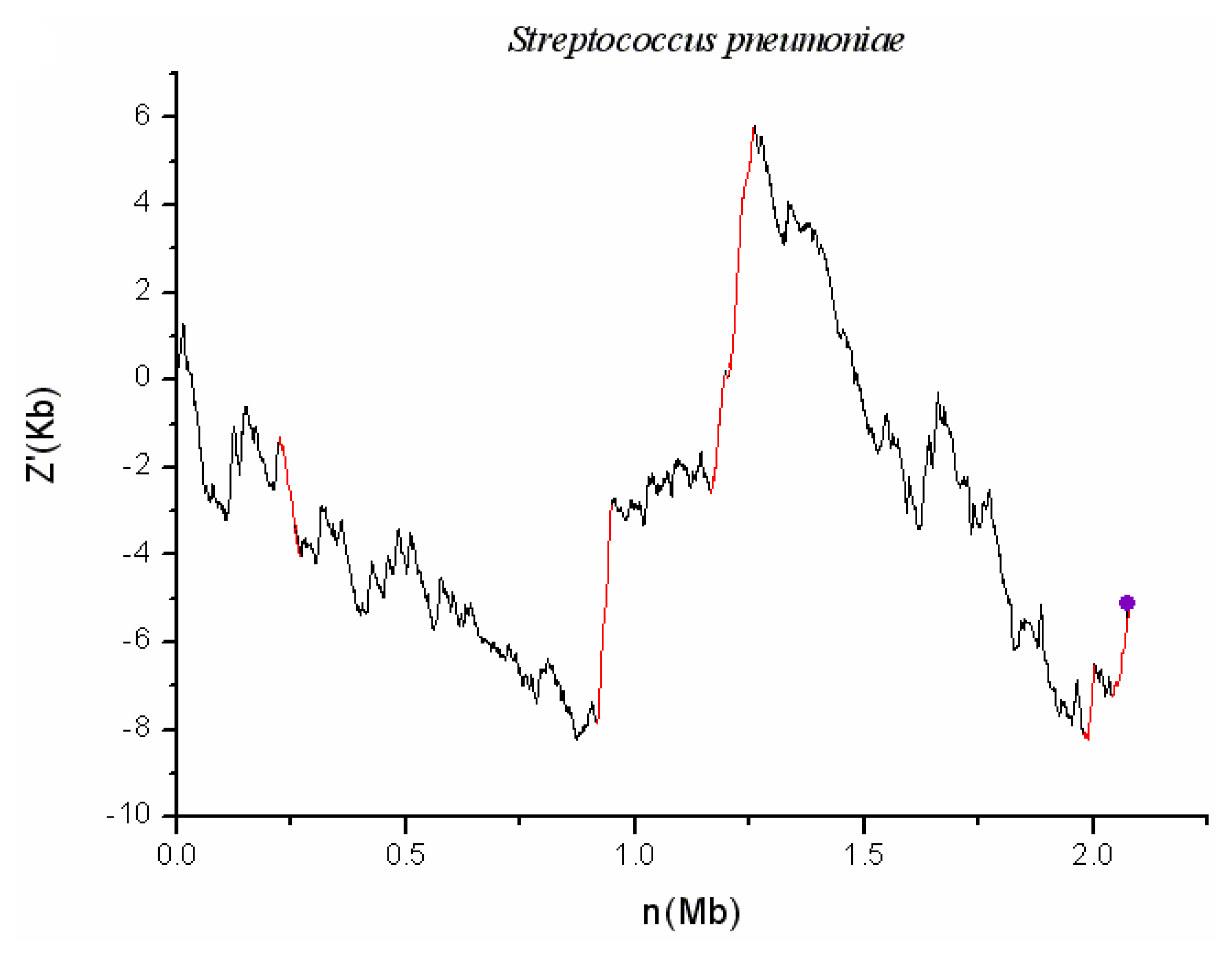
2.1. S. pneumoniae G54
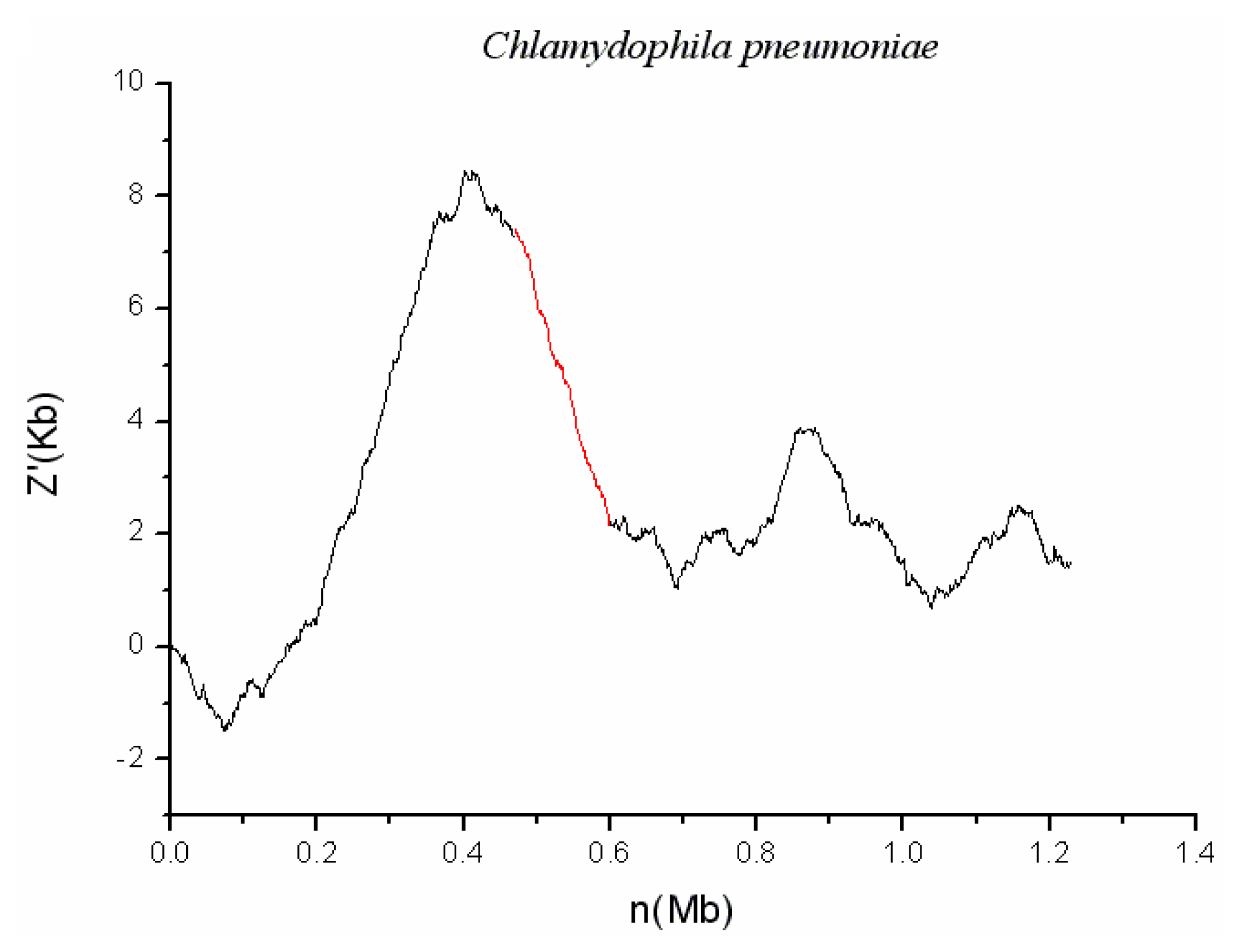
2.2. C. pneumoniae CWL029
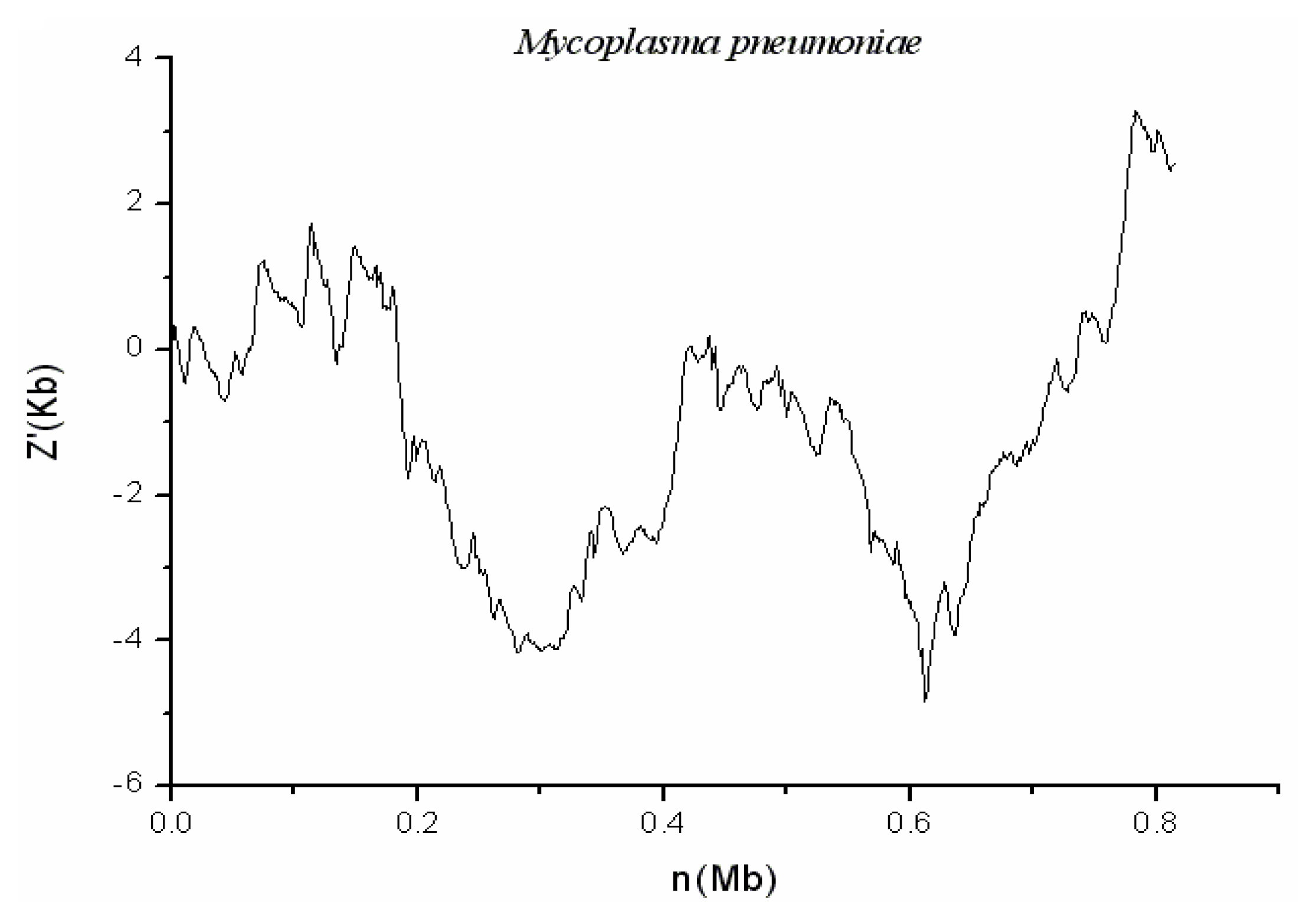
2.3. M. pneumoniae M129
3. Discussion
3.1. Conserved Features of Identified Genomic Islands
3.2. Comparison of the Cumulative GC Profile with SIGI Method
4. Experimental Section
4.1. Data Source
4.2. Cumulative GC Profile for Demonstrating GC Content Deviation of DNA Sequence
4.3. The h Index for Evaluating the Homogeneity of the GC Content of Genomic Island
4.4. BCN as an Index to Evaluate Codon Usage Bias
5. Conclusions
Acknowledgments
References
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar]
- Gogarten, J.P.; Townsend, J.P. Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol 2005, 3, 679–687. [Google Scholar]
- Hentschel, U.; Steinert, M.; Hacker, J. Common molecular mechanisms of symbiosis and pathogenesis. Trends Microbiol 2000, 8, 226–231. [Google Scholar]
- Do, J.H.; Miyano, S. The GC and window-averaged DNA curvature profile of secondary metabolite gene cluster in Aspergillus fumigatus genome. Appl. Microbiol. Biotechnol 2008, 80, 841–847. [Google Scholar]
- Hentschel, U.; Hacker, J. Pathogenicity islands: The tip of the iceberg. Microbes Infect 2001, 3, 545–548. [Google Scholar]
- Blum, G.; Ott, M.; Lischewski, A.; Ritter, A.; Imrich, H.; Tschape, H.; Hacker, J. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect. Immun 1994, 62, 606–614. [Google Scholar]
- Langille, M.G.; Hsiao, W.W.; Brinkman, F.S. Detecting genomic islands using bioinformatics approaches. Nat. Rev. Microbiol 2010, 8, 373–382. [Google Scholar]
- Greub, G.; Collyn, F.; Guy, L.; Roten, C.A. A genomic island present along the bacterial chromosome of the Parachlamydiaceae UWE25, an obligate amoebal endosymbiont, encodes a potentially functional F-like conjugative DNA transfer system. BMC Microbiol 2004, 4, 48. [Google Scholar]
- Karlin, S. Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol 2001, 9, 335–343. [Google Scholar]
- Zhang, C.T.; Wang, J.; Zhang, R. A novel method to calculate the G + C content of genomic DNA sequences. J. Biomol. Struct. Dyn 2001, 19, 333–341. [Google Scholar]
- Charkowski, A.O. Making sense of an alphabet soup: the use of a new bioinformatics tool for identification of novel gene islands. Focus on “identification of genomic islands in the genome of Bacillus cereus by comparative analysis with Bacillus anthracis”. Physiol. Genomics 2004, 16, 180–181. [Google Scholar]
- Zhang, C.T.; Zhang, R. Accurate localization of the integration sites of two genomic islands at single-nucleotide resolution in the genome of Bacillus cereus ATCC 10987. Comp. Funct. Genomics 2008. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, C.T. Genomic islands in the Corynebacterium efficiens genome. Appl. Environ. Microbiol 2005, 71, 3126–3130. [Google Scholar]
- Zhang, R.; Zhang, C.T. A systematic method to identify genomic islands and its applications in analyzing the genomes of Corynebacterium glutamicum and Vibrio vulnificus CMCP6 chromosome I. Bioinformatics 2004, 20, 612–622. [Google Scholar]
- Zhang, C.T.; Zhang, R. Genomic islands in Rhodopseudomonas palustris. Nat. Biotechnol 2004, 22, 1078–1079. [Google Scholar]
- Chen, L.L. Identification of genomic islands in six plant pathogens. Gene 2006, 374, 134–141. [Google Scholar]
- Ibrahim, Y.M.; Kerr, A.R.; McCluskey, J.; Mitchell, T.J. Role of htrA in the virulence and competence of Streptococcus pneumoniae. Infect. Immun 2004, 72, 3584–3591. [Google Scholar]
- Ragle, B.E.; Bubeck Wardenburg, J. Anti-α-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun 2009, 77, 2712–2718. [Google Scholar]
- Vernikos, G.S.; Parkhill, J. Resolving the structural features of genomic islands: A machine learning approach. Genome Res 2008, 18, 331–342. [Google Scholar]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden markov models. BMC Bioinformatics 2006, 7, 142. [Google Scholar]
- Langille, M.G.; Brinkman, F.S. IslandViewer. Available Online: http://www.pathogenomics.sfu.ca/islandviewer/query.php accessed on 10 May, 2010.
- National Center for Biotechnology Information. GenBank. 1982. Available Online: ftp://ftp.ncbi.nihgov/genbank/ accessed on 10 May, 2010.
- Guo, F.B.; Ou, H.Y.; Zhang, C.T. ZCURVE: A new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res 2003, 31, 1780–1789. [Google Scholar]
- Daubin, V.; Perrière, G. G + C3 structuring along the genome: A common feature in prokaryotes. Mol. Biol. Evol 2003, 20, 471–483. [Google Scholar]
- Cerdeno-Tárraga, A.M.; Efstratiou, A.; Dover, L.G.; Holden, M.T.; Pallen, M.; Bentley, S.D.; Besra, G.S.; Churcher, C.; James, K.D.; de Zoysa, A.; et al. The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res 2003, 31, 6516–6523. [Google Scholar]
- Khrustalev, V.V.; Barkovsky, E.V. “Protoisochores” in certain archaeal species are formed by replication-associated mutational pressure”. Biochimie 2011, 93, 160–167. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Segment | Start | End | h value | BCN | T (Transposase) or I (Integrase) | IS or Transposon |
|---|---|---|---|---|---|---|---|
| C. pneumoniae CWL029 | CPnGI01 | 470980 | 599631 | 0.034 | 18 | ||
| S. pneumoniae G54 | SPGGI01 | 137637 | 150199 | 0.048 | 21 | T (SPG_0139, SPG_0140) | |
| S. pneumoniae G54 | SPGGI02 | 224334 | 271125 | 0.021 | 20 | I (SPG_0281) | |
| S. pneumoniae G54 | SPGGI03 | 918880 | 947610 | 0.040 | 33 | T (SPG_0987) | |
| S. pneumoniae G54 | SPGGI04 | 1164899 | 1194336 | 0.057 | 21 | T (SPG_1195, SPG_1196, SPG_1200- SPG_1207) | Transposon (SPG_1225, SPG_1227, SPG_1228) IS (SPG_1196, SPG_1202-SPG_1207) |
| S. pneumoniae G54 | SPGGI05 | 1205234 | 1258720 | 0.083 | 37 | I (SPG_1258) T (SPG_1260) | Transposon (SPG_1242- SPG_1245, SPG_1250- SPG_1252, SPG_1259, SPG_1263-SPG_1267, SPG_1270-SPG_1273, SPG1275, SPG_1282, SPG_1285, SPG_1287- SPG_1293) |
| S. pneumoniae G54 | SPGGI06 | 1988635 | 2002624 | 0.011 | 31 | ||
| S. pneumoniae G54 | SPGGI07 | 2040970 | 2078937 | 0.077 | 21 | T (SPG_2157- SPG_2159) | IS (SPG_2157-SPG_2159) |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, F.-B.; Wei, W. Prediction of Genomic Islands in Three Bacterial Pathogens of Pneumonia. Int. J. Mol. Sci. 2012, 13, 3134-3144. https://doi.org/10.3390/ijms13033134
Guo F-B, Wei W. Prediction of Genomic Islands in Three Bacterial Pathogens of Pneumonia. International Journal of Molecular Sciences. 2012; 13(3):3134-3144. https://doi.org/10.3390/ijms13033134
Chicago/Turabian StyleGuo, Feng-Biao, and Wen Wei. 2012. "Prediction of Genomic Islands in Three Bacterial Pathogens of Pneumonia" International Journal of Molecular Sciences 13, no. 3: 3134-3144. https://doi.org/10.3390/ijms13033134