Use of the MLPA Assay in the Molecular Diagnosis of Gene Copy Number Alterations in Human Genetic Diseases

Abstract
:1. Background
2. Principles of MLPA Assay
3. MLPA Applications in Genetic Testing
3.1. MLPA and Neuromuscular Disorders
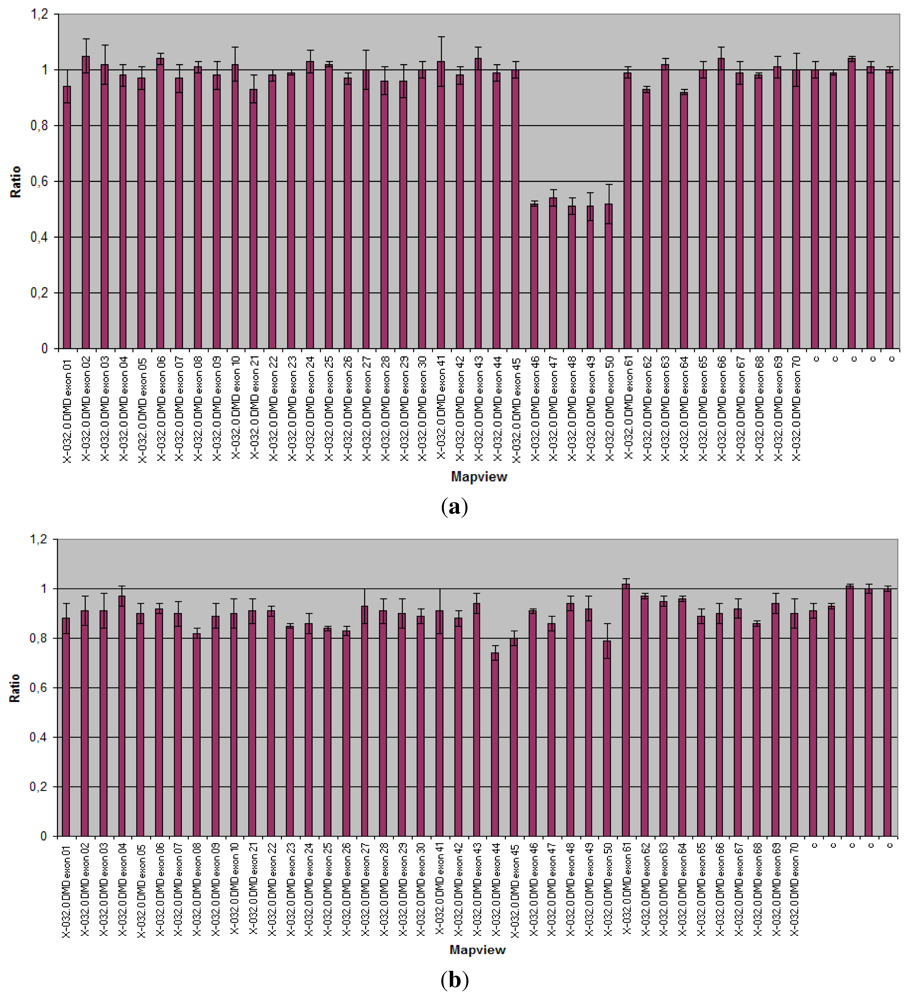
3.1.1. Dystrophinopathies
3.1.2. SMA
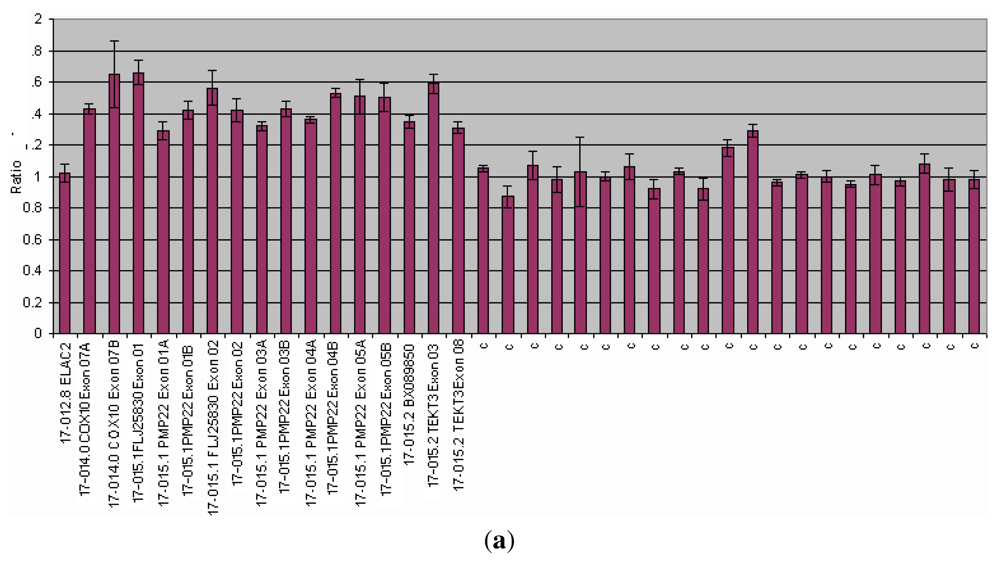
3.1.3. CMT and HNPP
3.2. MLPA and Analysis of the SHOX Gene
3.3. MLPA in Prenatal Diagnosis
3.4. MLPA and Cancer
3.4.1. MLPA and Hereditary Cancers
3.4.2. MLPA and Somatic Mutations in Cancer
3.5. MLPA and DNA Methylation
3.6. Limits of the MLPA Assay
4. Conclusions
References
- Armour, J.A.; Barton, D.E.; Cockburn, D.J.; Taylor, G.R. The detection of large deletions or duplications in genomic DNA. Hum. Mutat 2002, 20, 325–337. [Google Scholar]
- den Dunnen, J.T.; Grootscholten, P.M.; Bakker, E.; Blonden, L.A.; Ginjaar, H.B.; Wapenaar, M.C.; van Paassen, H.M.; van Broeckhoven, C.; Pearson, P.L.; van Ommen, G.J. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am. J. Hum. Genet 1989, 45, 835–847. [Google Scholar]
- van der Steege, G.; Grootscholten, P.M.; van der Vlies, P.; Draaijers, T.G.; Osinga, J.; Cobben, J.M.; Scheffer, H.; Buys, C.H. PCR based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995, 345, 985–986. [Google Scholar]
- Lee, C.; Iafrate, A.J.; Brothman, A.R. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat. Genet 2007, 39, 548–554. [Google Scholar]
- Choy, K.W.; Setlur, S.R.; Lee, C.; Lau, T.K. The impact of human copy number variation on a new era of genetic testing. BJOG 2010, 117, 391–398. [Google Scholar]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002, 30. [Google Scholar] [CrossRef]
- Kozlowski, P.; Jasinska, A.J.; Kwiatkowski, D.J. New applications and developments in the use of multiplex ligation-dependent probe amplification. Electrophoresis 2008, 23, 4627–3629. [Google Scholar]
- Jankowski, S.; Currie-Fraser, E.; Xu, L.; Coffa, J. Multiplex ligation-dependent probe amplification analysis on capillary electrophoresis instruments for a rapid gene copy number study. J. Biomol. Tech 2008, 9, 238–243. [Google Scholar]
- Coffa, J.; van de Wiel, M.A.; Diosdado, B.; Carvalho, B.; Schouten, J.; Meijer, G.A. MLPAnalyzer: Data analysis tool for reliable automated normalization of MLPA fragment data. Cell. Oncol 2008, 30, 323–335. [Google Scholar]
- Cáceres, A.; Armengol, L.; Villatoro, S.; González, J.R. MLPAstats: An R GUI package for the integrated analysis of copy number alterations using MLPA data. BMC Bioinform 2011, 12. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne Muscular Dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar]
- Forrest, S.M.; Cross, G.S.; Flint, T.; Speer, A.; Robson, K.J.; Davies, K.E. Further studies of gene deletions that cause Duchenne and Becker muscular dystrophies. Genomics 1988, 2, 109–114. [Google Scholar]
- Hu, X.Y.; Ray, P.N.; Murphy, E.G.; Thompson, M.W.; Worton, R.G. Duplicational mutation at the Duchenne muscular dystrophy locus: Its frequency, distribution, origin, and phenotype-genotype correlation. Am. J. Hum. Genet 1990, 46, 682–695. [Google Scholar]
- Roberts, R.G.; Bobrow, M.; Bentley, D.R. Point mutations in the dystrophin gene. Proc. Natl. Acad. Sci. USA 1992, 89, 2331–2335. [Google Scholar]
- Chamberlain, J.S.; Gibbs, R.A.; Ranier, J.E.; Nguyen, P.N.; Caskey, C.T. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 1988, 16, 11141–11156. [Google Scholar]
- Beggs, A.H.; Koenig, M.; Boyce, F.M.; Kunkel, L.M. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum. Genet 1990, 86, 45–48. [Google Scholar]
- Darras, B.T.; Harper, J.F.; Francke, U. Prenatal diagnosis and detection of carriers with DNA probes in Duchenne’s muscular dystrophy. N. Engl. J. Med 1987, 316, 985–992. [Google Scholar]
- Clemens, P.R.; Fenwick, R.G.; Chamberlain, J.S.; Gibbs, R.A.; de Andrade, M.; Chakraborty, R.; Caskey, C.T. Carrier detection and prenatal diagnosis in Duchenne and Becker muscular dystrophy families, using dinucleotide repeat polymorphisms. Am. J. Hum. Genet 1991, 49, 951–960. [Google Scholar]
- Prior, T.W.; Papp, A.C.; Snyder, P.J.; Highsmith, W.E., Jr; Friedman, K.J.; Perry, T.R.; Silverman, L.M.; Mendell, J.R. Determination of carrier status in Duchenne and Becker muscular dystrophies by quantitative polymerase chain reaction and allele-specific oligonucleotides. Clin. Chem. 1990, 36, 2113–2117. [Google Scholar]
- Abbs, S.; Bobrow, M. Report on the 16th ENMC Workshop—Carrier diagnosis of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord 1993, 3, 241–242. [Google Scholar]
- Voskova-Goldman, A.; Peier, A.; Caskey, C.T.; Richards, C.S.; Shaffer, L.G. DMD-specific FISH probes are diagnostically useful in the detection of female carriers of DMD gene deletions. Neurology 1997, 48, 1633–1638. [Google Scholar]
- Ligon, A.H.; Kashork, C.D.; Richards, C.S.; Shaffer, L.G. Identification of female carriers for Duchenne and Becker muscular dystrophies using a FISH-based approach. Eur. J. Hum. Genet 2000, 8, 293–298. [Google Scholar]
- Fortina, P.; Cheng, J.; Shoffner, M.A.; Surrey, S.; Hitchcock, W.M.; Kricka, L.J.; Wilding, P. Diagnosis of Duchenne/Becker muscular dystrophy and quantitative identification of carrier status by use of entangled solution capillary electrophoresis. Clin. Chem. 1997, 43, 745–751. [Google Scholar]
- Cinti, C.; Stuppia, L.; Maraldi, N.M. Combined use of PRINS and FISH in the study of the dystrophin gene. Am. J. Med. Genet 2002, 15, 115–118. [Google Scholar]
- White, S.; Kalf, M.; Liu, Q.; Villerius, M.; Engelsma, D.; Kriek, M.; Vollebregt, E.; Bakker, B.; van Ommen, G.J.; Breuning, M.H.; den Dunnen, J.T. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am. J. Hum. Genet 2002, 71, 365–374. [Google Scholar]
- Joncourt, F.; Neuhaus, B.; Jostarndt-Foegen, K.; Kleinle, S.; Steiner, B.; Gallati, S. Rapid identification of female carriers of DMD/BMD by quantitative real-time PCR. Hum. Mutat 2004, 23, 385–391. [Google Scholar]
- Hegde, M.R.; Chin, E.L.; Mulle, J.G.; Okou, D.T.; Warren, S.T.; Zwick, M.E. Microarray-based mutation detection in the dystrophin gene. Hum. Mutat 2008, 29, 1091–1099. [Google Scholar]
- del Gaudio, D.; Yang, Y.; Boggs, B.A.; Schmitt, E.S.; Lee, J.A.; Sahoo, T.; Pham, H.T.; Wiszniewska, J.; Chinault, A.C.; Beaudet, A.L.; et al. Molecular diagnosis of Duchenne/Becker muscular dystrophy: Enhanced detection of dystrophin gene rearrangements by oligonucleotide array-comparative genomic hybridization. Hum. Mutat 2008, 29, 1100–1107. [Google Scholar]
- Schwartz, M.; Dunø, M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Genet. Test 2004, 8, 361–367. [Google Scholar]
- Gatta, V.; Scarciolla, O.; Gaspari, A.R.; Palka, C.; de Angelis, M.V.; Di Muzio, A.; Guanciali-Franchi, P.; Calabrese, G.; Uncini, A.; Stuppia, L. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum. Genet 2005, 117, 92–98. [Google Scholar]
- Janssen, B.; Hartmann, C.; Scholz, V.; Jauch, A.; Zschocke, J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: Potential and pitfalls. Neurogenetics 2005, 6, 29–35. [Google Scholar]
- Lalic, T.; Vossen, R.H.; Coffa, J.; Schouten, J.P.; Guc-Scekic, M.; Radivojevic, D.; Djurisic, M.; Breuning, M.H.; White, S.J.; den Dunnen, J.T. Deletion and duplication screening in the DMD gene using MLPA. Eur. J. Hum. Genet 2005, 13, 1231–1234. [Google Scholar]
- Lai, K.; Lo, I.; Tong, T.; Cheng, L.; Lam, S. Detecting exon deletions and duplications of the DMD gene using Multiplex Ligation-dependent Probe Amplification (MLPA). Clin. Biochem 2006, 39, 367–372. [Google Scholar]
- Wang, Q.; Li-Ling, J.; Lin, C.; Wu, Y.; Sun, K.; Ma, H.; Jin, C. Characteristcs of dystrophin gene mutations among Chinese patients as revealed by multiplex ligation-dependent probe amplification. Genet. Test. Mol. Biomarkers 2009, 13, 23–30. [Google Scholar]
- Pikó, H.; Vancsó, V.; Nagy, B.; Bán, Z.; Herczegfalvi, A.; Karcagi, V. Dystrophin gene analysis in Hungarian Duchenne/Becker muscular dystrophy families—Detection of carrier status in symptomatic and asymptomatic female relatives. Neuromuscul. Disord 2009, 19, 108–112. [Google Scholar]
- Zeng, F.; Ren, Z.R.; Huang, S.Z.; Kalf, M.; Mommersteeg, M.; Smit, M.; White, S.; Jin, C.L.; Xu, M.; Zhou, D.W.; et al. Array-MLPA: Comprehensive detection of deletions and duplications and its application to DMD patients. Hum. Mutat 2008, 29, 190–197. [Google Scholar]
- Bunyan, D.J.; Skinner, A.C.; Ashton, E.J.; Sillibourne, J.; Brown, T.; Collins, A.L.; Cross, N.C.; Harvey, J.F.; Robinson, D.O. Simultaneous MLPA-based multiplex point mutation and deletion analysis of the dystrophin gene. Mol. Biotechnol 2007, 35, 135–140. [Google Scholar]
- Ogino, S.; Wilson, R.B. Spinal muscular atrophy: Molecular genetics and diagnostics. Expert Rev. Mol. Diagn 2004, 4, 15–29. [Google Scholar]
- Burghes, A.H. When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet 1997, 61, 9–10. [Google Scholar]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet 2002, 70, 358–368. [Google Scholar]
- Munsat, T.L.; Davies, K.E. International SMA consortium meeting (26–28 June 1992, Bonn, Germany). Neuromuscul. Disord 1992, 2, 423–428. [Google Scholar]
- McAndrew, P.E.; Parsons, D.W.; Simard, L.R.; Rochette, C.; Ray, P.N.; Mendell, J.R.; Prior, T.W.; Burghes, A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet 1997, 60, 1411–1422. [Google Scholar]
- van der Steege, G.; Grootscholten, P.M.; van der Vlies, P.; Draaijers, T.G.; Osinga, J.; Cobben, J.M.; Scheffer, H.; Buys, CH. PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995, 345, 985–986. [Google Scholar]
- Anhuf, D.; Eggermann, T.; Rudnik-Schoneborn, S.; Zerres, K. Determination of SMN1 and SMN2 copy number using TaqMan technology. Hum. Mutat 2003, 22, 74–78. [Google Scholar]
- Su, Y.N.; Hung, C.C.; Li, H.; Lee, C.N.; Cheng, W.F.; Tsao, P.N.; Chang, M.C.; Yu, C.L.; Hsieh, W.S.; Lin, W.L.; et al. Quantitative analysis of SMN1 and SMN2 genes based on DHPLC: A highly efficient and reliable carrier-screening test. Hum. Mutat 2005, 25, 460–467. [Google Scholar]
- Scarciolla, O.; Stuppia, L.; de Angelis, M.V.; Murru, S.; Palka, C.; Giuliani, R.; Pace, M.; di Muzio, A.; Torrente, I.; Morella, A.; et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 2006, 7, 269–276. [Google Scholar]
- Arkblad, E.L.; Darin, N.; Berg, K.; Kimber, E.; Brandberg, G.; Lindberg, C.; Holmberg, E.; Tulinius, M.; Nordling, M. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul. Disord 2006, 16, 830–838. [Google Scholar]
- Huang, C.H.; Chang, Y.Y.; Chen, C.H.; Kuo, Y.S.; Hwu, W.L.; Gerdes, T.; Ko, T.M. Copy number analysis of survival motor neuron genes by multiplex ligation-dependent probe amplification. Genet. Med 2007, 9, 241–248. [Google Scholar]
- Arkblad, E.; Tulinius, M.; Kroksmark, A.K.; Henricsson, M.; Darin, N. A population-based study of genotypic and phenotypic variability in children with spinal muscular atrophy. Acta Paediatr 2009, 98, 865–872. [Google Scholar]
- Yoon, S.; Lee, C.H.; Lee, K.A. Determination of SMN1 and SMN2 copy numbers in a Korean population using multiplex ligation-dependent probe amplification. Korean J. Lab. Med 2010, 30, 93–96. [Google Scholar]
- Petit, F.; Cuisset, J.M.; Rouaix-Emery, N.; Cancés, C.; Sablonnière, B.; Bieth, E.; Moerman, A.; Sukno, S.; Hardy, N.; Holder-Espinasse, M.; et al. Insights into genotype-phenotype correlations in spinal muscular atrophy: A retrospective study of 103 patients. Muscle Nerve 2011, 43, 26–30. [Google Scholar]
- Su, Y.N.; Hung, C.C.; Lin, S.Y.; Chen, F.Y.; Chern, J.P.; Tsai, C.; Chang, T.S.; Yang, C.C.; Li, H.; Ho, H.N.; et al. Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005–2009. A prospective population-based cohort study. PLoS One 2011, 5. [Google Scholar] [CrossRef]
- Passon, N.; de Wittenau, G.D.; Jurman, I.; Radovic, S.; Bregant, E.; Molinis, C.; Damante, G.; Lonigro, I.R. Quick MLPA test for quantification of SMN1 and SMN2 copy numbers. Mol. Cell. Probes 2010, 24, 310–314. [Google Scholar]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991, 66, 219–232. [Google Scholar]
- Chance, P.F.; Alderson, M.K.; Leppig, K.A.; Lensch, M.W.; Matsunami, N.; Smith, B.; Swanson, P.D.; Odelberg, S.J.; Disteche, C.M.; Bird, T.D. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993, 72, 143–151. [Google Scholar]
- Boerkoel, C.F.; Takashima, H.; Garcia, C.A.; Olney, R.K.; Johnson, J.; Berry, K.; Russo, P.; Kennedy, S.; Teebi, A.S.; Scavina, M.; et al. Charcot-Marie-Tooth disease and related neuropathies: Mutation distribution and genotype-phenotype correlation. Ann. Neurol 2002, 51, 190–201. [Google Scholar]
- Slater, H.; Bruno, D.; Ren, H.; La, P.; Burgess, T.; Hills, L.; Nouri, S.; Schouten, J.; Choo, K.H. Improved testing for CMT1A and HNPP using multiplex ligation-dependent probe amplification (MLPA) with rapid DNA preparations: Comparison with the interphase FISH method. Hum. Mutat 2004, 24, 164–171. [Google Scholar]
- Weterman, M.A.; van Ruissen, F.; de Wissel, M.; Bordewijk, L.; Samijn, J.P.; van der Pol, W.L.; Meggouh, F.; Baas, F. Copy number variation upstream of PMP22 in Charcot-Marie-Tooth disease. Eur. J. Hum. Genet 2010, 18, 421–428. [Google Scholar]
- Høyer, H.; Braathen, G.J.; Eek, A.K.; Skjelbred, C.F.; Russell, M.B. Charcot-Marie-Tooth caused by a copy number variation in myelin protein zero. Eur. J. Med. Genet 2011, 54, e580–e583. [Google Scholar]
- Rao, E.; Weiss, B.; Fukami, M.; Rump, A.; Niesler, B.; Mertz, A.; Muroya, K.; Binder, G.; Kirsch, S.; Winkelmann, M.; et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet 1997, 16, 54–63. [Google Scholar]
- Rappold, G.A.; Fukami, M.; Niesler, B.; Schiller, S.; Zumkeller, W.; Bettendorf, M.; Heinrich, U.; Vlachopapadoupoulou, E.; Reinehr, T.; Onigata, K.; et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J. Clin. Endocrinol. Metab 2002, 87, 1402–1406. [Google Scholar]
- Stuppia, L.; Calabrese, G.; Gatta, V.; Pintor, S.; Morizio, E.; Fantasia, D.; Franchi, P.G.; Rinaldi, M.M.; Scarano, G.; Concolino, D.; et al. SHOX mutations detected by FISH and direct sequencing in patients with short stature. J. Med. Genet 2003, 40. [Google Scholar] [CrossRef]
- Benito-Sanz, S.; Thomas, N.S.; Huber, C.; del Blanco, D.G.; Aza-Carmona, M.; Crolla, J.A.; Maloney, V.; Rappold, G.; Argente, J.; Campos-Barros, A.; et al. A novel class of Pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am. J. Hum. Genet 2005, 77, 533–544. [Google Scholar]
- Chen, J.; Wildhardt, G.; Zhong, Z.; Röth, R.; Weiss, B.; Steinberger, D.; Decker, J.; Blum, W.F.; Rappold, G. Enhancer deletions of the SHOX gene as a frequent cause of short stature: The essential role of a 250 kb downstream regulatory domain. J. Med. Genet 2009, 46, 834–839. [Google Scholar]
- Niesler, B.; Röth, R.; Wilke, S.; Fujimura, F.; Fischer, C.; Rappold, G. The novel human SHOX allelic variant database. Hum. Mutat 2007, 28, 933–938. [Google Scholar]
- Blum, W.F.; Crowe, B.J.; Quigley, C.A.; Jung, H.; Cao, D.; Ross, J.L.; Braun, L.; Rappold, G. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: Two-year results of a randomized, controlled, multicenter trial. J. Clin. Endocrinol. Metab 2007, 92, 219–228. [Google Scholar]
- Scalco, R.C.; Melo, S.S.; Pugliese-Pires, P.N.; Funari, M.F.; Nishi, M.Y.; Arnhold, I.J.; Mendonca, B.B.; Jorge, A.A. Effectiveness of the combined recombinant human growth hormone and gonadotropin-releasing hormone analog therapy in pubertal patients with short stature due to SHOX deficiency. J. Clin. Endocrinol. Metab 2010, 95, 328–332. [Google Scholar]
- Gatta, V.; Antonucci, I.; Morizio, E.; Palka, C.; Fischetto, R.; Mokini, V.; Tumini, S.; Calabrese, G.; Stuppia, L. Identification and characterization of different SHOX gene deletions in patients with Leri-Weill dyschondrosteosys by MLPA assay. J. Hum. Genet 2007, 52, 21–27. [Google Scholar]
- Funari, M.F.; Jorge, A.A.; Pinto, E.M.; Arnhold, I.J.; Mendonca, B.B.; Nishi, M.Y. Cryptic intragenic deletion of the SHOX gene in a family with Léri-Weill dyschondrosteosis detected by Multiplex Ligation-Dependent Probe Amplification (MLPA). Arq. Bras. Endocrinol. Metabol 2008, 52, 1382–1387. [Google Scholar]
- Fukami, M.; Dateki, S.; Kato, F.; Hasegawa, Y.; Mochizuki, H.; Horikawa, R.; Ogata, T. Identification and characterization of cryptic SHOX intragenic deletions in three Japanese patients with Léri-Weill dyschondrosteosis. J. Hum. Genet 2008, 53, 454–459. [Google Scholar]
- Funari, M.F.; Jorge, A.A.; Souza, S.C.; Billerbeck, A.E.; Arnhold, I.J.; Mendonca, B.B.; Nishi, M.Y. Usefulness of MLPA in the detection of SHOX deletions. Eur. J. Med. Genet 2010, 53, 234–238. [Google Scholar]
- Benito-Sanz, S.; Barroso, E.; Heine-Suñer, D.; Hisado-Oliva, A.; Romanelli, V.; Rosell, J.; Aragones, A.; Caimari, M.; Argente, J.; Ross, J.L.; et al. Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J. Clin. Endocrinol. Metab 2011, 96, E404–E412. [Google Scholar]
- Slater, H.R.; Bruno, D.L.; Ren, H.; Pertile, M.; Schouten, J.P.; Choo, K.H. Rapid, high throughput prenatal detection of aneuploidy using a novel quantitative method (MLPA). J. Med. Genet 2003, 40, 907–912. [Google Scholar]
- Gerdes, T.; Kirchhoff, M.; Lind, A.M.; Larsen, G.V.; Schwartz, M.; Lundsteen, C. Computer-assisted prenatal aneuploidy screening for chromosome 13, 18, 21, X and Y based on multiplex ligation-dependent probe amplification (MLPA). Eur. J. Hum. Genet 2005, 13, 171–175. [Google Scholar]
- van Opstal, D.; Boter, M.; de Jong, D.; van den Berg, C.; Brüggenwirth, H.T.; Wildschut, H.I.; de Klein, A.; Galjaard, R.J. Rapid aneuploidy detection with multiplex ligation-dependent probe amplification: A prospective study of 4000 amniotic fluid samples. Eur. J. Hum. Genet 2009, 17, 112–121. [Google Scholar]
- Guo, Q.; Zhou, Y.; Wang, X.; Li, Q. Simultaneous detection of trisomies 13, 18, and 21 with multiplex ligation-dependent probe amplification-based real-time PCR. Clin. Chem 2010, 56, 1451–1459. [Google Scholar]
- Yan, J.B.; Xu, M.; Xiong, C.; Zhou, D.W.; Ren, Z.R.; Huang, Y.; Mommersteeg, M.; van Beuningen, R.; Wang, Y.T.; Liao, S.X.; et al. Rapid screening for chromosomal aneuploidies using array-MLPA. BMC Med. Genet 2011, 12. [Google Scholar] [CrossRef]
- Mazoyer, S. Genomic rearrangements in the BRCA1 and BRCA2 genes. Hum. Mutat 2005, 25, 415–422. [Google Scholar]
- Hogervorst, F.B.; Nederlof, P.M.; Gille, J.J.; McElgunn, C.J.; Grippeling, M.; Pruntel, R.; Regnerus, R.; van Welsem, T.; van Spaendonk, R.; Menko, F.H.; et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res 2003, 63, 1449–1453. [Google Scholar]
- Montagna, M.; Palma, M.D.; Menin, C.; Agata, S.; de Nicolo, A.; Chieco-Bianchi, L.; D’Andrea, E. Genomic rearrangements account for more than one-third of the BRCA1 mutations in northern Italian breast/ovarian cancer families. Hum. Mol. Genet 2003, 12, 1055–1061. [Google Scholar]
- Belogianni, I.; Apessos, A.; Mihalatos, M.; Razi, E.; Labropoulos, S.; Petounis, A.; Gaki, V.; Keramopoulos, A.; Pandis, N.; Kyriacou, K.; et al. Characterization of a novel large deletion and single point mutations in the BRCA1 gene in a Greek cohort of families with suspected hereditary breast cancer. BMC Cancer 2004, 7. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, C.; John, A.L.; Klaes, R.; Hofmann, W.; Bielen, R.; Koehler, R.; Janssen, B.; Bartram, C.R.; Arnold, N.; Zschocke, J. Large BRCA1 gene deletions are found in 3% of German high-risk breast cancer families. Hum. Mutat 2004, 24. [Google Scholar] [CrossRef]
- Woodward, A.M.; Davis, T.A.; Silva, A.G.; Kirk, J.A.; Leary, J.A. kConFab Investigators. Large genomic rearrangements of both BRCA2 and BRCA1 are a feature of the inherited breast/ovarian cancer phenotype in selected families. J. Med. Genet 2005, 42. [Google Scholar] [CrossRef]
- Veschi, S.; Aceto, G.; Scioletti, A.P.; Gatta, V.; Palka, G.; Cama, A.; Mariani-Costantini, R.; Battista, P.; Calò, V.; Barbera, F.; et al. High prevalence of BRCA1 deletions in BRCAPRO-positive patients with high carrier probability. Ann. Oncol 2007, 18. [Google Scholar]
- Bunyan, D.J.; Eccles, D.M.; Sillibourne, J.; Wilkins, E.; Thomas, N.S.; Shea-Simonds, J.; Duncan, P.J.; Curtis, C.E.; Robinson, D.O.; Harvey, J.F.; et al. Dosage analysis of cancer predisposition genes by multiplex ligation-dependent probe amplification. Br. J. Cancer 2004, 91, 1155–1159. [Google Scholar]
- Michils, G.; Tejpar, S.; Thoelen, R.; van Cutsem, E.; Vermeesch, J.R.; Fryns, J.P.; Legius, E.; Matthijs, G. Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): A Belgian study. Hum. Mutat 2005, 25, 125–134. [Google Scholar]
- McCart, A.; Latchford, A.; Volikos, E.; Rowan, A.; Tomlinson, I.; Silver, A. A novel exon duplication event leading to a truncating germ-line mutation of the APC gene in a familial adenomatous polyposis family. Fam. Cancer 2006, 5, 205–208. [Google Scholar]
- Pagenstecher, C.; Gadzicki, D.; Stienen, D.; Uhlhaas, S.; Mangold, E.; Rahner, N.; Arslan-Kirchner, M.; Propping, P.; Friedl, W.; Aretz, S. A complex rearrangement in the APC gene uncovered by multiplex ligation-dependent probe amplification. J. Mol. Diagn 2007, 9, 122–126. [Google Scholar]
- Nakagawa, H.; Hampel, H.; de la Chapelle, A. Identification and characterization of genomic rearrangements of MSH2 and MLH1 in Lynch syndrome (HNPCC) by novel techniques. Hum. Mutat 2003, 22. [Google Scholar] [CrossRef]
- Taylor, C.F.; Charlton, R.S.; Burn, J.; Sheridan, E.; Taylor, GR. Genomic deletions in MSH2 or MLH1 are a frequent cause of hereditary non-polyposis colorectal cancer: Identification of novel and recurrent deletions by MLPA. Hum. Mutat. 2003, 22, 428–433. [Google Scholar]
- Wang, Y.; Friedl, W.; Lamberti, C.; Jungck, M.; Mathiak, M.; Pagenstecher, C.; Propping, P.; Mangold, E. Hereditary nonpolyposis colorectal cancer: Frequent occurrence of large genomic deletions in MSH2 and MLH1 genes. Int. J. Cancer 2003, 103, 636–641. [Google Scholar]
- Ainsworth, P.J.; Koscinski, D.; Fraser, B.P.; Stuart, J.A. Family cancer histories predictive of a high risk of hereditary non-polyposis colorectal cancer associate significantly with a genomic rearrangement in hMSH2 or hMLH1. Clin. Genet 2004, 66, 183–188. [Google Scholar]
- Baudhuin, L.M.; Mai, M.; French, A.J.; Kruckeberg, K.E.; Swanson, R.L.; Winters, J.L.; Courteau, L.K.; Thibodeau, S.N. Analysis of hMLH1 and hMSH2 gene dosage alterations in hereditary nonpolyposis colorectal cancer patients by novel methods. J. Mol. Diagn 2005, 7, 226–235. [Google Scholar]
- Grabowski, M.; Mueller-Koch, Y.; Grasbon-Frodl, E.; Koehler, U.; Keller, G.; Vogelsang, H.; Dietmaier, W.; Kopp, R.; Siebers, U.; Schmitt, W.; et al. Deletions account for 17% of pathogenic germline alterations in MLH1 and MSH2 in hereditary nonpolyposis colorectal cancer (HNPCC) families. Genet. Test 2005, 9, 138–146. [Google Scholar]
- de Lellis, L.; Curia, M.C.; Catalano, T.; de Toffol, S.; Bassi, C.; Mareni, C.; Bertario, L.; Battista, P.; Mariani-Costantini, R.; Radice, P.; et al. Combined use of MLPA and nonfluorescent multiplex PCR analysis by high performance liquid chromatography for the detection of genomic rearrangements. Hum. Mutat 2006, 27, 1047–1056. [Google Scholar]
- Wehner, M.; Mangold, E.; Sengteller, M.; Friedrichs, N.; Aretz, S.; Friedl, W.; Propping, P.; Pagenstecher, C. Hereditary nonpolyposis colorectal cancer: Pitfalls in deletion screening in MSH2 and MLH1 genes. Eur. J. Hum. Genet 2005, 13, 983–986. [Google Scholar]
- van Dijk, M.C.; Rombout, P.D.; Boots-Sprenger, S.H.; Straatman, H.; Bernsen, M.R.; Ruiter, D.J.; Jeuken, J.W. Multiplex ligation-dependent probe amplification for the detection of chromosomal gains and losses in formalin-fixed tissue. Diagn. Mol. Pathol 2005, 14, 9–16. [Google Scholar]
- Jeuken, J.; Cornelissen, S.; Boots-Sprenger, S.; Gijsen, S.; Wesseling, P. Multiplex ligation-dependent probe amplification: A diagnostic tool for simultaneous identification of different genetic markers in glial tumors. J. Mol. Diagn 2006, 8, 433–443. [Google Scholar]
- Franco-Hernandez, C.; Martinez-Glez, V.; Alonso, M.E.; de Campos, J.M.; Isla, A.; Vaquero, J.; Gutierrez, M.; Rey, J.A. Gene dosage and mutational analyses of EGFR in oligodendrogliomas. Int. J. Oncol 2007, 30, 209–215. [Google Scholar]
- Nowee, M.E.; Snijders, A.M.; Rockx, D.A.; de Wit, R.M.; Kosma, V.M.; Hämäläinen, K.; Schouten, J.P.; Verheijen, R.H.; van Diest, P.J.; Albertson, D.G.; et al. DNA profiling of primary serous ovarian and fallopian tube carcinomas with array comparative genomic hybridization and multiplex ligation-dependent probe amplification. J. Pathol 2007, 213, 46–55. [Google Scholar]
- Owens, M.; Ellard, S.; Vaidya, B. Analysis of gross deletions in the MEN1 gene in patients with multiple endocrine neoplasia type 1. Clin. Endocrinol. (Oxford) 2008, 68, 350–354. [Google Scholar]
- Villamón, E.; Piqueras, M.; Mackintosh, C.; Alonso, J.; de Alava, E.; Navarro, S.; Noguera, R. Comparison of different techniques for the detection of genetic risk-identifying chromosomal gains and losses in neuroblastoma. Virchows Arch 2008, 453, 47–55. [Google Scholar]
- Ewald, C.; Hofmann, T.; Kuhn, S.A.; Deufel, T.; Beetz, C.; Kalff, R. Methylation-specific multiplex ligation-dependent probe amplification in meningiomas. J. Neurooncol 2008, 90, 267–273. [Google Scholar]
- Guervós, M.A.; Marcos, C.A.; Llorente, J.L.; Nuño, A.S.; Suárez, C.; Hermsen, M. Genetic differences between primary larynx and pharynx carcinomas and their matched lymph node metastases by multiplex ligation-dependent probe amplification. Oral Oncol 2009, 45, 600–604. [Google Scholar]
- Vignoli, M.; Scaini, M.C.; Ghiorzo, P.; Sestini, R.; Bruno, W.; Menin, C.; Gensini, F.; Piazzini, M.; Testori, A.; Manoukian, S.; et al. Genomic rearrangements of the CDKN2A locus are infrequent in Italian malignant melanoma families without evidence of CDKN2A/CDK4 point mutations. Melanoma Res 2008, 18, 431–437. [Google Scholar]
- Damato, B.; Dopierala, J.; Klaasen, A.; van Dijk, M.; Sibbring, J.; Coupland, S.E. Multiplex ligation-dependent probe amplification of uveal melanoma: Correlation with metastatic death. Invest. Ophthalmol. Vis. Sci 2009, 50, 3048–3055. [Google Scholar]
- Damato, B.; Dopierala, J.A.; Coupland, S.E. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin. Cancer Res 2010, 16, 6083–6092. [Google Scholar]
- Franco-Hernández, C.; Martínez-Glez, V.; de Campos, J.M.; Isla, A.; Vaquero, J.; Gutiérrez, M.; Casartelli, C.; Rey, J.A. Allelic status of 1p and 19q in oligodendrogliomas and glioblastomas: Multiplex ligation-dependent probe amplification versus loss of heterozygosity. Cancer Genet. Cytogenet 2009, 190, 93–96. [Google Scholar]
- Buffart, T.E.; van Grieken, N.C.; Tijssen, M.; Coffa, J.; Ylstra, B.; Grabsch, H.I.; van de Velde, C.J.; Carvalho, B.; Meijer, G.A. High resolution analysis of DNA copy-number aberrations of chromosomes 8, 13, and 20 in gastric cancers. Virchows Arch 2009, 455, 213–223. [Google Scholar]
- Zhang, D.; Wang, Z.; Luo, Y.; Xu, Y.; Liu, Y.; Yang, W.; Zhang, X. Analysis of DNA copy number aberrations by multiple ligation-dependent probe amplification on 50 intestinal type gastric cancers. J. Surg. Oncol 2010, 103, 124–132. [Google Scholar]
- Tepeli, E.; Muslumanoglu, M.H.; Uludag, A.; Buyukpinarbasili, N.; Ozdemir, M.; Oznur, M.; Aslan, H.; Artan, S. Detection of deletions and/or amplifications of genes related with lung cancer by multiplex ligation-dependent probe amplification (MLPA) technique. Cancer Biol. Ther 2009, 8, 2160–2165. [Google Scholar]
- Masson, D.; Rioux-Leclercq, N.; Fergelot, P.; Jouan, F.; Mottier, S.; Théoleyre, S.; Bach-Ngohou, K.; Patard, J.J.; Denis, M.G. Loss of expression of TIMP3 in clear cell renal cell carcinoma. Eur. J. Cancer 2010, 46, 1430–1437. [Google Scholar]
- Buijs, A.; Krijtenburg, P.J.; Meijer, E. Detection of risk-identifying chromosomal abnormalities and genomic profiling by multiplex ligation-dependent probe amplification in chronic lymphocytic leukemia. Haematologica 2006, 91, 1434–1435. [Google Scholar]
- Coll-Mulet, L.; Santidrián, A.F.; Cosialls, A.M.; Iglesias-Serret, D.; de Frias, M.; Grau, J.; Menoyo, A.; González-Barca, E.; Pons, G.; Domingo, A.; et al. Multiplex ligation-dependent probe amplification for detection of genomic alterations in chronic lymphocytic leukaemia. Br. J. Haematol 2008, 142, 793–801. [Google Scholar]
- Stevens-Kroef, M.; Simons, A.; Gorissen, H.; Feuth, T.; Weghuis, D.O.; Buijs, A.; Raymakers, R.; van Kessel, A.G. Identification of chromosomal abnormalities relevant to prognosis in chronic lymphocytic leukemia using multiplex ligation-dependent probe amplification. Cancer Genet. Cytogenet 2009, 195, 97–104. [Google Scholar]
- Abdool, A.; Donahue, A.C.; Wohlgemuth, J.G.; Yeh, C.H. Detection, analysis and clinical validation of chromosomal aberrations by multiplex ligation-dependent probe amplification in chronic leukemia. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Al Zaabi, E.A.; Fernandez, L.A.; Sadek, I.A.; Riddell, D.C.; Greer, W.L. Multiplex ligation-dependent probe amplification versus multiprobe fluorescence in situ hybridization to detect genomic aberrations in chronic lymphocytic leukemia: A tertiary center experience. J. Mol. Diagn 2010, 12, 197–203. [Google Scholar]
- Fabris, S.; Scarciolla, O.; Morabito, F.; Cifarelli, R.A.; Dininno, C.; Cutrona, G.; Matis, S.; Recchia, A.G.; Gentile, M.; Ciceri, G.; et al. Multiplex ligation-dependent probe amplification and fluorescence in situ hybridization to detect chromosomal abnormalities in chronic lymphocytic leukemia: A comparative study. Genes Chromosomes Cancer 2011, 50, 726–734. [Google Scholar]
- Schwab, C.J.; Jones, L.R.; Morrison, H.; Ryan, S.L.; Yigittop, H.; Schouten, J.P.; Harrison, C.J. Evaluation of multiplex ligation-dependent probe amplification as a method for the detection of copy number abnormalities in B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer 2010, 49, 1104–1113. [Google Scholar]
- Donahue, A.C.; Abdool, A.K.; Gaur, R.; Wohlgemuth, J.G.; Yeh, C.H. Multiplex ligation-dependent probe amplification for detection of chromosomal abnormalities in myelodysplastic syndrome and acute myeloid leukemia. Leuk. Res 2011, 35, 1477–1483. [Google Scholar]
- Purnomosari, D.; Aryandono, T.; Setiaji, K.; Nugraha, S.B.; Pals, G.; van Diest, P.J. Comparison of multiplex ligation dependent probe amplification to immunohistochemistry for assessing HER-2/neu amplification in invasive breast cancer. Biotech. Histochem 2006, 81, 79–85. [Google Scholar]
- Moerland, E.; van Hezik, R.L.; van der Aa, T.C.; van Beek, M.W.; van den Brule, A.J. Detection of HER2 amplification in breast carcinomas: Comparison of Multiplex Ligation-dependent Probe Amplification (MLPA) and Fluorescence In Situ Hybridization (FISH) combined with automated spot counting. Cell. Oncol 2006, 28, 151–159. [Google Scholar]
- Moelans, C.B.; de Weger, R.A.; van Blokland, M.T.; Ezendam, C.; Elshof, S.; Tilanus, M.G.; van Diest, P.J. HER-2/neu amplification testing in breast cancer by multiplex ligation-dependent probe amplification in comparison with immunohistochemistry and in situ hybridization. Cell. Oncol 2009, 31, 1–10. [Google Scholar]
- Moelans, C.B.; de Weger, R.A.; Ezendam, C.; van Diest, P.J. HER-2/neu amplification testing in breast cancer by Multiplex Ligation-dependent Probe Amplification: Influence of manual- and laser microdissection. BMC Cancer 2009, 9. [Google Scholar] [CrossRef]
- Moelans, C.B.; de Weger, R.A.; van Blokland, M.T.; van der Wall, E.; van Diest, P.J. Simultaneous detection of TOP2A and HER2 gene amplification by multiplex ligation-dependent probe amplification in breast cancer. Mod. Pathol 2010, 23, 62–70. [Google Scholar]
- Paulsen, M.; Ferguson-Smith, A.C. DNA methylation in genomic imprinting, development, and disease. J. Pathol 2001, 195, 97–110. [Google Scholar]
- Liang, G.; Salem, C.E.; Yu, M.C.; Nguyen, H.D.; Gonzales, F.A.; Nguyen, T.T.; Nichols, P.W.; Jones, P.A. DNA methylation differences associated with tumor tissues identified by genome scanning analysis. Genomics 1998, 53, 260–268. [Google Scholar]
- Nygren, A.O.H.; Ameziane, N.; Duarte, H.; Vijzelaar1, R.; Waisfisz, Q.; Hess, C.; Schouten, J.P.; Errami, A. Methylation-Specific MLPA (MS-MLPA): Simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res 2005, 33. [Google Scholar] [CrossRef]
- Cassidy, S.B.; Driscoll, D.J. Prader-Willi syndrome. Eur. J. Hum. Genet 2009, 17, 3–13. [Google Scholar]
- Shao, H.; Lip, V.; Wu, B.L. Effectiveness of multiplex ligation-dependent probe amplification assay used for detecting deletion of Prader-Willi syndrome. Beijing Da Xue Xue Bao 2005, 37, 64–67. [Google Scholar]
- Procter, M.; Chou, L.S.; Tang, W.; Jama, M.; Mao, R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin. Chem 2006, 52, 1276–1283. [Google Scholar]
- Bittel, D.C.; Kibiryeva, N.; Butler, M.G. Methylation-specific multiplex ligation-dependent probe amplification analysis of subjects with chromosome 15 abnormalities. Genet. Test 2007, 11, 467–475. [Google Scholar]
- Dikow, N.; Nygren, A.O.; Schouten, J.P.; Hartmann, C.; Krämer, N.; Janssen, B.; Zschocke, J. Quantification of the methylation status of the PWS/AS imprinted region: Comparison of two approaches based on bisulfite sequencing and methylation-sensitive MLPA. Mol. Cell. Probes 2007, 21, 208–215. [Google Scholar]
- Priolo, M.; Sparago, A.; Mammì, C.; Cerrato, F.; Laganà, C.; Riccio, A. MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur. J. Hum. Genet 2008, 16, 565–571. [Google Scholar]
- Scott, R.H.; Douglas, J.; Baskcomb, L.; Nygren, A.O.; Birch, J.M.; Cole, T.R.; Cormier-Daire, V.; Eastwood, D.M.; Garcia-Minaur, S.; Lupunzina, P.; et al. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) robustly detects and distinguishes 11p15 abnormalities associated with overgrowth and growth retardation. J. Med. Genet 2008, 45, 106–113. [Google Scholar]
- Eggermann, T.; Schönherr, N.; Eggermann, K.; Buiting, K.; Ranke, M.B.; Wollmann, H.A.; Binder, G. Use of multiplex ligation-dependent probe amplification increases the detection rate for 11p15 epigenetic alterations in Silver-Russell syndrome. Clin. Genet 2008, 73, 79–84. [Google Scholar]
- Zeschnigk, M.; Albrecht, B.; Buiting, K.; Kanber, D.; Eggermann, T.; Binder, G.; Gromoll, J.; Prott, E.C.; Seland, S.; Horsthemke, B. IGF2/H19 hypomethylation in silver-russell syndrome and isolated hemihypoplasia. Eur. J. Hum. Genet 2008, 16, 328–334. [Google Scholar]
- Jeuken, J.W.; Cornelissen, S.J.; Vriezen, M.; Dekkers, M.M.; Errami, A.; Sijben, A.; Boots-Sprenger, S.H.; Wesseling, P. MS-MLPA: An attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab. Invest 2007, 87, 1055–1065. [Google Scholar]
- Erlandson, A.; Appelqvist, F.; Enerbäck, C. Epigenetic mutations in CDKN2A in western Swedish families with hereditary malignant melanoma. Mol. Med. Rep 2008, 1, 89–91. [Google Scholar]
- van Nifterik, K.A.; van den Berg, J.; van der Meide, W.F.; Ameziane, N.; Wedekind, L.E.; Steenbergen, R.D.; Leenstra, S.; Lafleur, M.V.; Slotman, B.J.; Stalpers, L.J.; et al. Absence of the MGMT protein as well as methylation of the MGMT promoter predict the sensitivity for temozolomide. Br. J. Cancer 2010, 103, 29–35. [Google Scholar]
- Park, C.K.; Kim, J.; Yim, S.Y.; Lee, A.R.; Han, J.H.; Kim, C.Y.; Park, S.H.; Kim, T.M.; Lee, S.H.; Choi, S.H.; et al. Usefulness of MS-MLPA for detection of MGMT promoter methylation in the evaluation of pseudoprogression in glioblastoma patients. Neuro Oncol 2011, 13, 195–202. [Google Scholar]
- Lee, J.Y.; Park, C.K.; Park, S.H.; Wang, K.C.; Cho, B.K.; Kim, S.K. MGMT promoter gene methylation in pediatric glioblastoma: Analysis using MS-MLPA. Child’s Nerv. Syst 2011, 27, 1877–1883. [Google Scholar]
- Chen, K.; Sawhney, R.; Khan, M.; Benninger, M.S.; Hou, Z.; Sethi, S.; Stephen, J.K.; Worsham, M.J. Methylation of multiple genes as diagnostic and therapeutic markers in primary head and neck squamous cell carcinoma. Arch. Otolaryngol. Head Neck Surg 2007, 133, 1131–1138. [Google Scholar]
- Berkhout, M.; Nagtegaal, I.D.; Cornelissen, S.J.; Dekkers, M.M.; van de Molengraft, F.J.; Peters, W.H.; Nagengast, F.M.; van Krieken, J.H.; Jeuken, J.W. Chromosomal and methylation alterations in sporadic and familial adenomatous polyposis-related duodenal carcinomas. Mod. Pathol 2007, 20, 1253–1262. [Google Scholar]
- Hess, C.J.; Errami, A.; Berkhof, J.; Denkers, F.; Ossenkoppele, G.J.; Nygren, A.O.; Schuurhuis, G.J.; Waisfisz, Q. Concurrent methylation of promoters from tumor associated genes predicts outcome in acute myeloid leukemia. Leuk. Lymphoma 2008, 49, 1132–1141. [Google Scholar]
- Hess, C.J.; Ameziane, N.; Schuurhuis, G.J.; Errami, A.; Denkers, F.; Kaspers, G.J.; Cloos, J.; Joenje, H.; Reinhardt, D.; Ossenkoppele, G.J.; et al. Hypermethylation of the FANCC and FANCL promoter regions in sporadic acute leukaemia. Cell. Oncol 2008, 30, 299–306. [Google Scholar]
- Buyru, N.; Altinisik, J.; Ozdemir, F.; Demokan, S.; Dalay, N. Methylation profiles in breast cancer. Cancer Invest 2009, 27, 307–312. [Google Scholar]
- Berginc, G.; Bracko, M.; Glavac, D. MS-MLPA reveals progressive age-dependent promoter methylation of tumor suppressor genes and possible role of IGSF4 gene in colorectal carcinogenesis of microsatellite instable tumors. Cancer Invest 2010, 28, 94–102. [Google Scholar]
- Marzese, D.M.; Gago, F.E.; Vargas-Roig, L.M.; Roqué, M. Simultaneous analysis of the methylation profile of 26 cancer related regions in invasive breast carcinomas by MS-MLPA and drMS-MLPA. Mol. Cell. Probes 2010, 24, 271–280. [Google Scholar]
- Castro, M.; Grau, L.; Puerta, P.; Gimenez, L.; Venditti, J.; Quadrelli, S.; Sánchez-Carbayo, M. Multiplexed methylation profiles of tumor suppressor genes and clinical outcome in lung cancer. J. Transl. Med 2010, 8. [Google Scholar] [CrossRef]
- Eberth, S.; Schneider, B.; Rosenwald, A.; Hartmann, E.M.; Romani, J.; Zaborski, M.; Siebert, R.; Drexler, H.G.; Quentmeier, H. Epigenetic regulation of CD44 in Hodgkin and non-Hodgkin lymphoma. BMC Cancer 2010, 10. [Google Scholar] [CrossRef]
- Zauber, N.P.; Denehy, T.R.; Taylor, R.R.; Ongcapin, E.H.; Marotta, S.P.; Sabbath-Solitare, M.; Kulkarni, R.; Pradhan, T.S.; Hermelin, D.; Bishop, D.T. Microsatellite instability and DNA methylation of endometrial tumors and clinical features in young women compared with older women. Int. J. Gynecol. Cancer 2010, 20, 1549–1556. [Google Scholar]
- Suijkerbuijk, K.P.; Pan, X.; van der Wall, E.; van Diest, P.J.; Vooijs, M. Comparison of different promoter methylation assays in breast cancer. Anal. Cell Pathol. (Amst.) 2010, 33, 133–141. [Google Scholar]
- Moelans, C.B.; Verschuur-Maes, A.H.; van Diest, P.J. Frequent promoter hypermethylation of BRCA2, CDH13, MSH6, PAX5, PAX6 and WT1 in ductal carcinoma in situ and invasive breast cancer. J. Pathol 2011, 225, 222–231. [Google Scholar]
- Agundez, M.; Grau, L.; Palou, J.; Algaba, F.; Villavicencio, H.; Sanchez-Carbayo, M. Evaluation of the methylation status of tumour suppressor genes for predicting bacillus Calmette-Guérin response in patients with T1G3 high-risk bladder tumours. Eur. Urol 2011, 60, 131–140. [Google Scholar]
- Valencia, A.; Cervera, J.; Such, E.; Ibañez, M.; Gómez, I.; Luna, I.; Senent, L.; Oltra, S.; Sanz, M.A.; Sanz, G.F. Aberrant methylation of tumor suppressor genes in patients with refractory anemia with ring sideroblasts. Leuk. Res 2011, 35, 479–483. [Google Scholar]
- Cabello, M.J.; Grau, L.; Franco, N.; Orenes, E.; Alvarez, M.; Blanca, A.; Heredero, O.; Palacios, A.; Urrutia, M.; Fernández, J.M.; et al. Multiplexed methylation profiles of tumor suppressor genes in bladder cancer. J. Mol. Diagn. 2011, 13, 29–40. [Google Scholar]
- Stephen, J.K.; Chitale, D.; Narra, V.; Chen, K.M.; Sawhney, R.; Worsham, M.J. DNA methylation in thyroid tumorigenesis. Cancers 2011, 3, 1732–1743. [Google Scholar]
- Chen, K.M.; Stephen, J.K.; Raju, U.; Worsham, M.J. Delineating an epigenetic continuum for initiation, transformation and progression to breast cancer. Cancers 2011, 3, 1580–1592. [Google Scholar]
- Leong, K.J.; Wei, W.; Tannahill, L.A.; Caldwell, G.M.; Jones, C.E.; Morton, D.G.; Matthews, G.M.; Bach, S.P. Methylation profiling of rectal cancer identifies novel markers of early-stage disease. Br. J. Surg 2011, 98, 724–734. [Google Scholar]
- Pavicic, W.; Perkiö, E.; Kaur, S.; Peltomäki, P. Altered methylation at MicroRNA-associated CpG islands in hereditary and sporadic carcinomas: A Methylation-Specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA)-based approach. Mol. Med 2011, 17, 726–735. [Google Scholar]
- Bunyan, D.J.; Skinner, A.C.; Ashton, E.J.; Sillibourne, J.; Brown, T.; Collins, A.L.; Cross, N.C.; Harvey, J.F.; Robinson, D.O. Simultaneous MLPA-based multiplex point mutation and deletion analysis of the dystrophin gene. Mol. Biotechnol 2007, 35, 135–140. [Google Scholar]
- Stern, R.F.; Roberts, R.G.; Mann, K.; Yau, S.C.; Berg, J.; Ogilvie, CM. Multiplex ligation-dependent probe amplification using a completely synthetic probe set. Biotechniques 2004, 37, 399–405. [Google Scholar]
- Erlandson, A.; Samuelsson, L.; Hagberg, B.; Kyllerman, M.; Vujic, M.; Wahlstrom, J. Multiplex ligation-dependent probeamplification (MLPA) detects large deletions in the MECP2 gene of Swedish Rett syndrome patients. Genet. Test 2003, 7, 329–332. [Google Scholar]
- Colosimo, A.; Gatta, V.; Guida, V.; Leodori, E.; Foglietta, E.; Rinaldi, S.; Cappabianca, M.P.; Amato, A.; Stuppia, L.; Dallapiccola, B. Application of MLPA assay to characterize unsolved α-globin gene rearrangements. Blood. Cells Mol. Dis 2011, 46, 139–144. [Google Scholar]
- Kipp, B.R.; Roellinger, S.E.; Lundquist, P.A.; Highsmith, W.E.; Dawson, D.B. Development and clinical implementation of a combination deletion PCR and multiplex ligation-dependent probe amplification assay for detecting deletions involving the human α-globin gene cluster. J. Mol. Diagn 2011, 13, 549–557. [Google Scholar]
- Barbaro, M.; Cools, M.; Looijenga, L.H.; Drop, S.L.; Wedell, A. Partial deletion of the NR5A1 (SF1) gene detected by synthetic probe MLPA in a patient with XY gonadal disorder of sex development. Sex. Dev 2011, 5, 181–187. [Google Scholar]
- Jang, J.H.; Jin, D.K.; Kim, J.H.; Tan, H.K.; Kim, J.W.; Lee, S.Y.; Ki, C.S.; Park, H.D. Multiplex ligation-dependent probe amplification assay for diagnosis of congenital adrenal hyperplasia. Ann. Clin. Lab. Sci 2011, 41, 44–47. [Google Scholar]
- Rooms, L.; Vandeweyer, G.; Reyniers, E.; van Mol, K.; de Canck, I.; van der Aa, N.; Rossau, R.; Kooy, R.F. Array-based MLPA to detect recurrent copy number variations in patients with idiopathic mental retardation. Am. J. Med. Genet. A 2011, 155A, 343–348. [Google Scholar]
- Scarciolla, O.; Brancati, F.; Valente, E.M.; Ferraris, A.; de Angelis, M.V.; Valbonesi, S.; Garavaglia, B.; Uncini, A.; Palka, G.; Stuppia, L.; et al. Multiplex ligation-dependent probe amplification assay for simultaneous detection of Parkinson’s disease gene rearrangements. Mov. Disord 2007, 22, 2274–2278. [Google Scholar]
- Pankratz, N.; Dumitriu, A.; Hetrick, K.N.; Sun, M.; Latourelle, J.C.; Wilk, J.B.; Halter, C.; Doheny, K.F.; Gusella, J.F.; Nichols, W.C.; et al. PSG-PROGENI and GenePD investigators, coordinators and molecular genetic laboratories. Copy number variation in familial parkinson disease. PLoS One 2011, 6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Method | Advantages | Disadvantages |
|---|---|---|
| MLPA | Detects small rearrangements Up to 40 targets High throughput Low cost | Cannot detect copy neutral loss of heterozygosity. May have problems with mosaicism, tumor heterogeneity, or contamination with normal cells. |
| FISH | Detects balanced rearrangements Detects mosaicism Detects tumor heterogeneity Can quantify multiple copies | Cannot detect copy neutral loss of heterozygosity. Cannot detect small rearrangements (e.g., deletions <100 kb or duplications >500 kb). Limited number of targets and throughput. |
| Quantitative/Sq-PCR | Detects small rearrangements and even point mutations Can quantify multiple copies Low cost | Test optimization and efficiency is a concern. Limited number of targets. May have problems with mosaicism, tumor heterogeneity, or contamination with normal cells. |
| Southern blot | Detects small rearrangements Detects mosaicism | Cannot detect copy neutral loss of heterozygosity. Not quantitative. Laborious and time consuming Limited number of targets and throughput. |
| CGH array | Can detect very small rearrangements Can probe entire genome Low cost per data point | Cannot detect copy neutral loss of heterozygosity. Costly equipment and reagents Low throughput |
| SNP array | Can detect copy neutral loss or heterozygosity Can probe entire genome Low cost per data point | Cannot detect small rearrangements (e.g., deletions or duplications <100 kb). Costly equipment and reagents Low throughput |
| Disease | Gene | Application | MLPA Advantages | Proportion of Cases Due to del/dupl | MLPA Detection Rate of del/dupl | References |
|---|---|---|---|---|---|---|
| DMD/BMD | DMD gene | Diagnosis | All the 79 exons of the DMD-gene analyzed in two reactions. Detection of duplications and heterozygous deletions. | 60 70% del 5 10% dupl | >99% | [25,29 37] |
| SMA | SMN1-SMN2 | Diagnosis | Detection of heterozygous SMN1 loss. Ability to discriminate between SMN1 deletions and conversions to SMN2. | 95% | >98% | [46 51] |
| CMT/HNPP | PMP22 | Diagnosis | All the PMP22 exons analyzed in a single reaction. A single probe set able to analyze two different conditions. | Dupl 70% 80% CMT. Deletion 85% of HNPP cases | >95% | [57 59] |
| LWD | SHOX | Diagnosis/Research | Ability to analyze both the SHOX gene coding region and the enhancer region. Detection of partial gene deletions and duplications. | 40% del | >80% | [68 72] |
| Aneuplodies of 13, 18, 21, X and Y chromosomes | - | Diagnosis | A single probe mix for the detection of several aneuploidies. | - | >95% | [73 76] |
| BC, OC | BRCA1, BRCA2 | Diagnosis/Research | Detection of large gene rearrangements. | 15 30% | NA | [79 84] |
| FAP | APC | Diagnosis/Research | Detection of large gene rearrangements. | 15 25% | NA | [85 89] |
| HNPCC | MLH1, MSH2 | Diagnosis/Research | A single probe set for all exons of both genes | MLH1:5% MSH2: 20% | NA | [90 95] |
| PWS/AS | 15q11-q13 region | Diagnosis | A single MS-MLPA probe set used for the analysis of both diseases. | Methylation abnormality 95% | 99% | [130 133] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stuppia, L.; Antonucci, I.; Palka, G.; Gatta, V. Use of the MLPA Assay in the Molecular Diagnosis of Gene Copy Number Alterations in Human Genetic Diseases. Int. J. Mol. Sci. 2012, 13, 3245-3276. https://doi.org/10.3390/ijms13033245
Stuppia L, Antonucci I, Palka G, Gatta V. Use of the MLPA Assay in the Molecular Diagnosis of Gene Copy Number Alterations in Human Genetic Diseases. International Journal of Molecular Sciences. 2012; 13(3):3245-3276. https://doi.org/10.3390/ijms13033245
Chicago/Turabian StyleStuppia, Liborio, Ivana Antonucci, Giandomenico Palka, and Valentina Gatta. 2012. "Use of the MLPA Assay in the Molecular Diagnosis of Gene Copy Number Alterations in Human Genetic Diseases" International Journal of Molecular Sciences 13, no. 3: 3245-3276. https://doi.org/10.3390/ijms13033245