Prediction of Bioluminescent Proteins Using Auto Covariance Transformation of Evolutional Profiles

Abstract
:1. Introduction
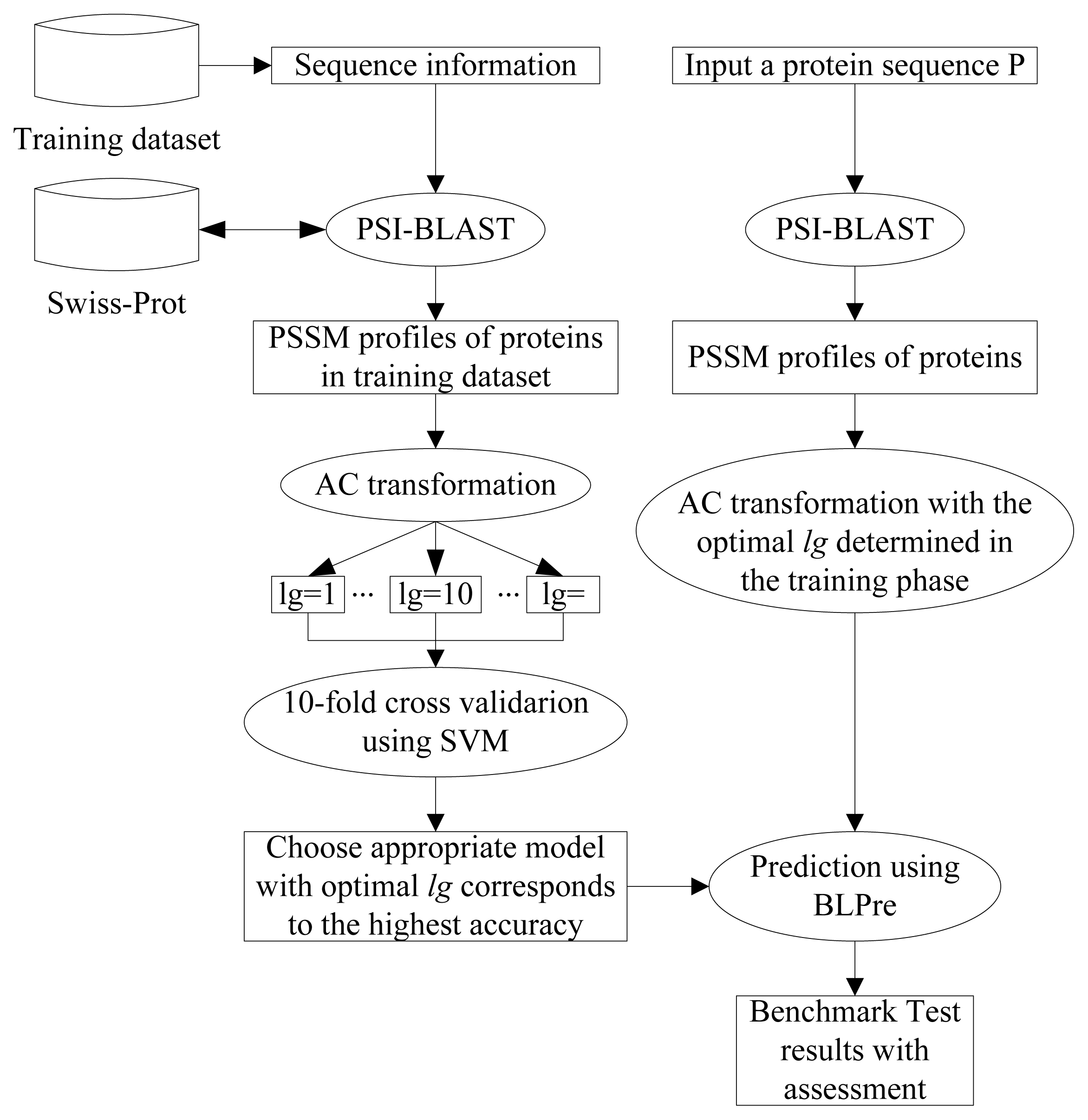
2. Materials and Methods
2.1. Datasets
2.2. Position Specific Scoring Matrix
2.3. Auto Covariance
2.4. Support Vector Machine
2.5. Model Construction
2.6. Performance Evaluation
3. Results and Discussion
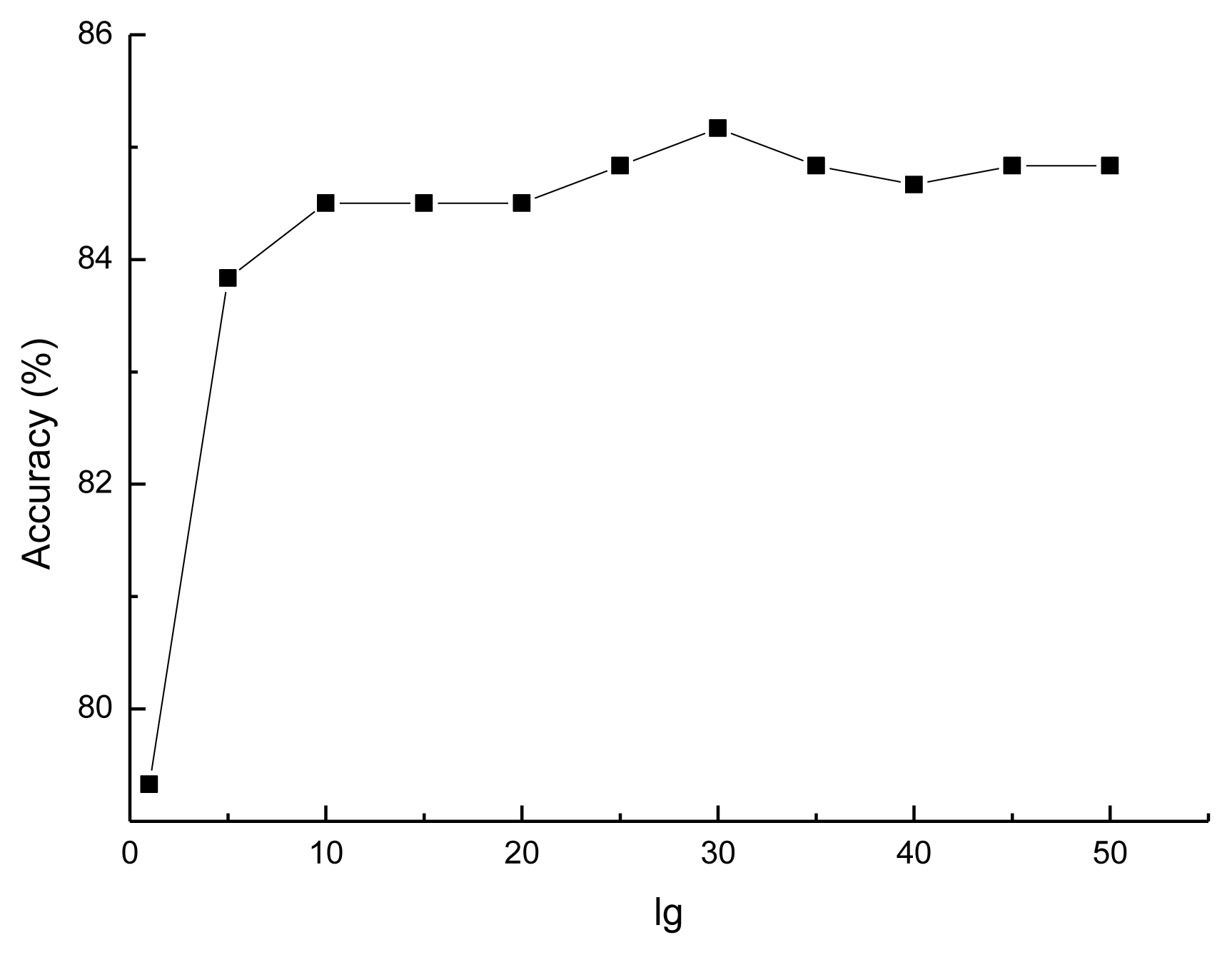
3.1. Selecting the Optimal lg for the Prediction Model
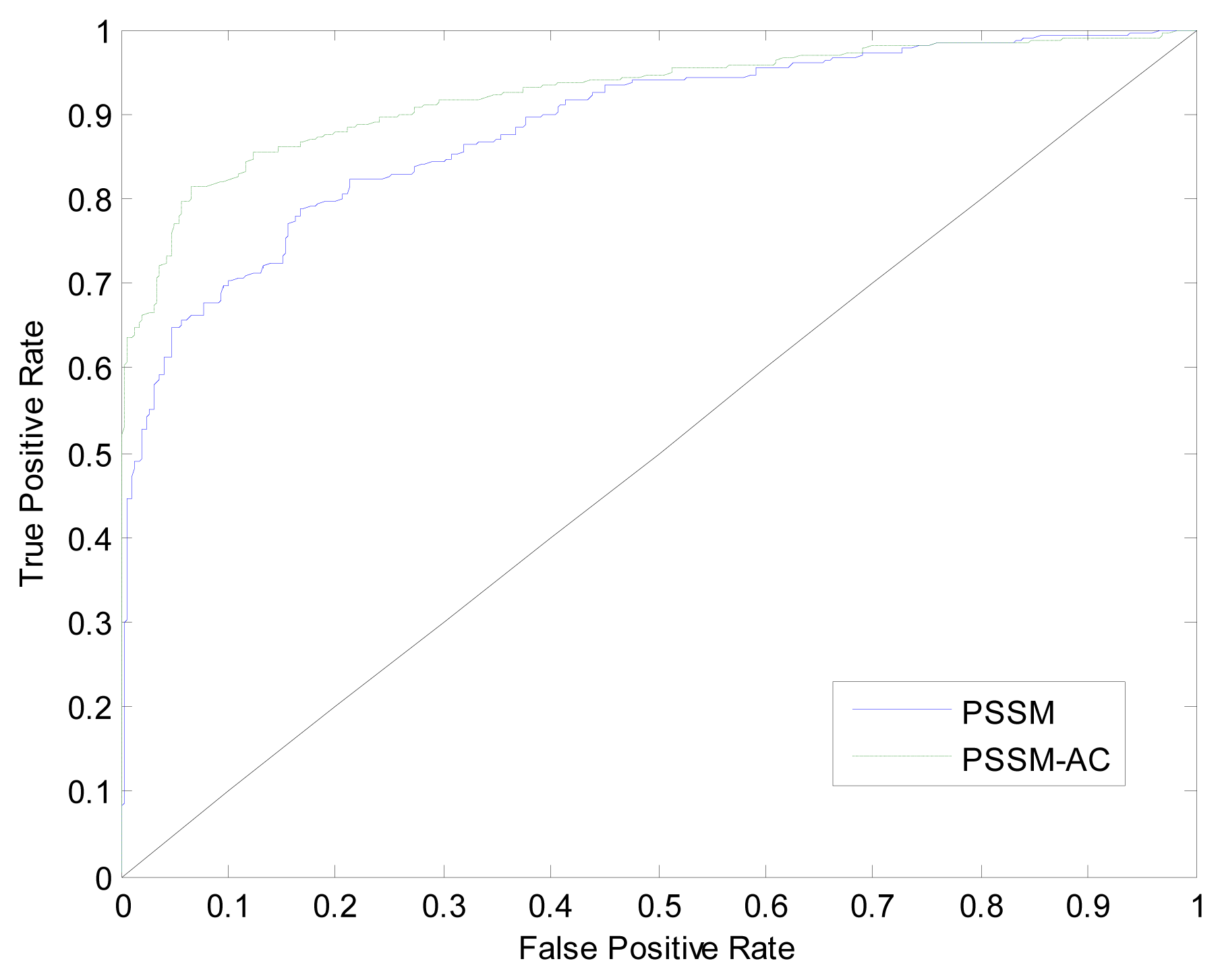
3.2. Comparison with Simple PSI-BLAST Search Method
3.3. Comparison with Other Methods
4. Conclusions
Acknowledgments
References
- Hastings, J.W. Bioluminescence; Academic Press: New York NY, USA, 1995. [Google Scholar]
- Wilson, T. Comments on the mechanisms of chemi- and bioluminescence. Photochem. Photobiol 1995, 62, 601–606. [Google Scholar]
- Haddock, S.H.D.; Moline, M.A.; Case, J.F. Bioluminescence in the Sea. Ann. Rev. Mar. Sci 2010, 2, 293–343. [Google Scholar]
- Lloyd, J.E. Insect Bioluminescence; Academic Press: New York NY, USA, 1978. [Google Scholar]
- White, E.H.; Rapaport, E.; Seliger, H.H.; Hopkins, T.A. The chemi- and bioluminescence of firefly luciferin: An efficient chemical production of electronically excited states. Bioorg. Chem 1971, 1, 92–122. [Google Scholar]
- Shimomura, O.; Johnson, F.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, aequorea. J. Cell. Phys 1962, 59, 223–239. [Google Scholar]
- Pierre, A.V.; Val, J.W. Fluorescent and bioluminescent protein-fragment complementation assays in the study of G protein-coupled receptor oligomerization and signaling. Mol. Pharmacol 2009, 75, 733–739. [Google Scholar]
- Kandaswamy, K.K.; Ganesan, P.; Mehrnaz, K.H.; Kai, K.; Martinetz, T. BLProt: Prediction of bioluminescent proteins based on Support Vector Machine and ReliefF feature selection. BMC Bioinforma 2011, 12. [Google Scholar] [CrossRef]
- Kawashima, S.; Ogata, H.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res 1999, 27, 368–369. [Google Scholar]
- Liu, T.G.; Geng, X.B.; Zheng, X.Q.; Li, R.S.; Wang, J. Accurate prediction of protein structural class using auto covariance transformation of PSI-BLAST profiles. Amino Acids 2011. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.Z.; Xiao, R.Q.; Zeng, Y.H.; Xiao, J.M.; Tan, F.Y.; Li, M.L. Using auto covariance method for functional discrimination of membrance proteins based on evolution information. Amino Acids 2010, 38, 1497–1503. [Google Scholar]
- BLPre. Available online: http://59.73.198.144/AFP_PSSM/ accessed on 10 February 2012.
- BLProt dataset. Available online: http://www.inb.uni-luebeck.de/tools-demos/bioluminescent%20protein/BLProt accessed on 23 December 2011.
- Chou, K.C.; Shen, H.B. Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Chou, K.C.; Wu, Z.C.; Xiao, X. iLoc-Euk: A multi-lable classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinformatics 2007, 8. [Google Scholar] [CrossRef]
- Song, J.; Burrage, K.; Yuan, Z.; Huber, T. Prediction of cis/trans isomerization in proteins using PSI-BLAST profiles and secondary structure information. BMC Bioinformatics 2006, 7. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 2007, 23, 538–544. [Google Scholar]
- Biswas, A.K.; Noman, N.; Sikder, A.R. Machine learning approach to predict protein phosphorylation sites by incorporating evolutionary information. BMC Bioinformatics 2010, 11. [Google Scholar] [CrossRef]
- Ruchi, V.; Grish, C.V.; Raghava, G.P.S. Prediction of mitochondrial proteins of malaria parasite using split amino acid composition and PSSM profile. Amino Acids 2010, 39, 101–110. [Google Scholar]
- Zhao, X.W.; Li, X.T.; Ma, Z.Q.; Yin, M.H. Prediction of lysine ubiquitylation with ensemble classifier and feature selection. Int. J. Mol. Sci 2011, 12, 8347–8361. [Google Scholar]
- Altschul, S.; Wootton, J.; Gertz, E.; Agarwala, R.; Morgulis, A.; Schaffer, A.; Yu, Y. Protein database searches using compositionally adjusted substitution matrices. FEBS J 2005, 272, 5101–5109. [Google Scholar]
- Altschul, S.; Madden, T.; Schaffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Schaffer, A.; Aravind, L.; Madden, T.; Shavirin, S.; Spouge, J.; Wolf, Y.; Koonin, E.; Altschul, S. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 2001, 29, 2994–3005. [Google Scholar]
- Shen, H.B.; Chou, K.C. Predicting protein fold pattern with functional domain and sequential evolution information. J. Theor. Biol 2009, 256, 441–446. [Google Scholar]
- Shen, H.B.; Chou, K.C. Quatldent: A web server for identifying protein quaternary structural attribute by fusing functional domain and sequential evolution information. J. Proteome Res 2009, 8, 1577–1584. [Google Scholar]
- Song, J.; Yuan, Z.; Tan, H.; Huber, T.; Burrage, K. Predicting disulfide connectivity from protein sequence using multiple sequence feature vectors and secondary structure. Bioinformatics 2007, 23, 3147–3154. [Google Scholar]
- Zhu, L.; Yang, J.; Song, J.N.; Chou, K.C.; Shen, H.B. Improving the accuracy of predicting disulfide connectivity by feature selection. J. Comput. Chem 2010, 31, 1478–1485. [Google Scholar]
- Song, J.; Tan, H.; Takemoto, K.; Akutsu, T. HSEpred: Predict half-sphere exposure from protein sequence. Bioinformatics 2008, 24, 1489–1497. [Google Scholar]
- Lobley, A.; Sadowski, M.I.; Jones, D.T. pGenTHREADER and pDomTHERADER: New methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 2009, 25, 1761–1767. [Google Scholar]
- Chauhan, J.S.; Mishra, N.K.; Raghava, G.P. Identification of ATP binding residues of a protein from its primary sequence. BMC Bioinformatics 2009, 10. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, H.; Chen, K.; Shen, S.; Ruan, J.; Kurgan, L. Accurate sequence-based prediction of catalytic residues. Bioinformatics 2008, 24, 2329–2338. [Google Scholar]
- Wold, S.; Jonsson, J.; Sjostrom, M.; Rannar, S. DNA and peptide sequences and chemical processes multivariately modeled by principal component analysis and partial least squares projection to latent structures. Anal. Chim. Acta 1993, 277, 239–253. [Google Scholar]
- Guo, Y.; Li, M.; Lu, M.; Wen, Z.; Huang, Z. Predicting G-protein coupled receptors-G-protein coupling specificity based on autocross-covariance transform. Proteins 2006, 65, 55–60. [Google Scholar]
- Guo, Y.Z.; Yu, L.Z.; Wen, Z.N.; Li, M.L. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res 2008, 36, 3025–3030. [Google Scholar]
- Dong, Q.W.; Zhou, S.G.; Guan, J.H. A new taxonomy-based protein folds recognition approach based on autocross-covariance transformation. Bioinformatics 2009, 25, 2655–2662. [Google Scholar]
- Wu, J.; Li, M.; Yu, L.; Wang, C. An ensemble classifier of support vector machines used to predict protein structural classes by fusing auto covariance and pseudo-amino acid composition. Protein J 2010, 29, 62–67. [Google Scholar]
- Zeng, Y.H.; Guo, Y.Z.; Xiao, R.Q.; Yang, L.; Yu, L.Z.; Li, M.L. Using the augmented Chou’s pseudo amino acid composition for predicting protein submitochondria locations based on auto covariance approach. J. Theor. Biol 2009, 259, 366–372. [Google Scholar]
- Liu, T.G.; Zheng, X.Q.; Wang, C.H.; Wang, J. Prediction of subcellular location of apoptosis proteins using pseudo amino acid composition: An approach from auto covariance transformation. Protein Pept. Lett 2010, 17, 1263–1269. [Google Scholar]
- Khan, A.; Javed, S.J. Predicting regularities in lattice constants of GdfeO3-type perovskites. Acta Crystallogr 2008, B64, 120–122. [Google Scholar]
- Qiu, J.D.; Huang, J.H.; Liang, R.P.; Lu, X.Q. Prediction of G-protein-coupled receptors based on the concept of Chou’s pseudo amino acid composition: An approach from discrete wavelet transform. Anal. Biochem 2009, 390, 68–73. [Google Scholar]
- Zhang, S.; Ding, S.; Wang, T. High-accuracy prediction of protein structural class for low-similarity sequences based on predicted secondary structure. Biochimie 2011, 4, 710–714. [Google Scholar]
- Vapnik, V. Statistical Learning Theory; Wiley-Interscience: New York, NY, USA, 1998. [Google Scholar]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machine. ACM Trans. Intell. Syst. Technol 2011, 2, 1–27. [Google Scholar]
- Chou, K.C.; Zhang, C.T. Review: Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol 1995, 30, 275–349. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc 2008, 3, 153–162. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Sn (%) | Sp (%) | AC (%) |
|---|---|---|---|
| PSSM-400 | 72.00 | 86.33 | 79.32 |
| PSSM-AC | 79.33 | 91.00 | 85.17 |
| BLProt [8] | 74.47 | 84.21 | 80.00 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhao, X.; Li, J.; Huang, Y.; Ma, Z.; Yin, M. Prediction of Bioluminescent Proteins Using Auto Covariance Transformation of Evolutional Profiles. Int. J. Mol. Sci. 2012, 13, 3650-3660. https://doi.org/10.3390/ijms13033650
Zhao X, Li J, Huang Y, Ma Z, Yin M. Prediction of Bioluminescent Proteins Using Auto Covariance Transformation of Evolutional Profiles. International Journal of Molecular Sciences. 2012; 13(3):3650-3660. https://doi.org/10.3390/ijms13033650
Chicago/Turabian StyleZhao, Xiaowei, Jiakui Li, Yanxin Huang, Zhiqiang Ma, and Minghao Yin. 2012. "Prediction of Bioluminescent Proteins Using Auto Covariance Transformation of Evolutional Profiles" International Journal of Molecular Sciences 13, no. 3: 3650-3660. https://doi.org/10.3390/ijms13033650