Genome-Wide DNA Methylation Changes between the Superficial and Deep Backfat Tissues of the Pig

Abstract
:
1. Introduction
2. Results and Discussion
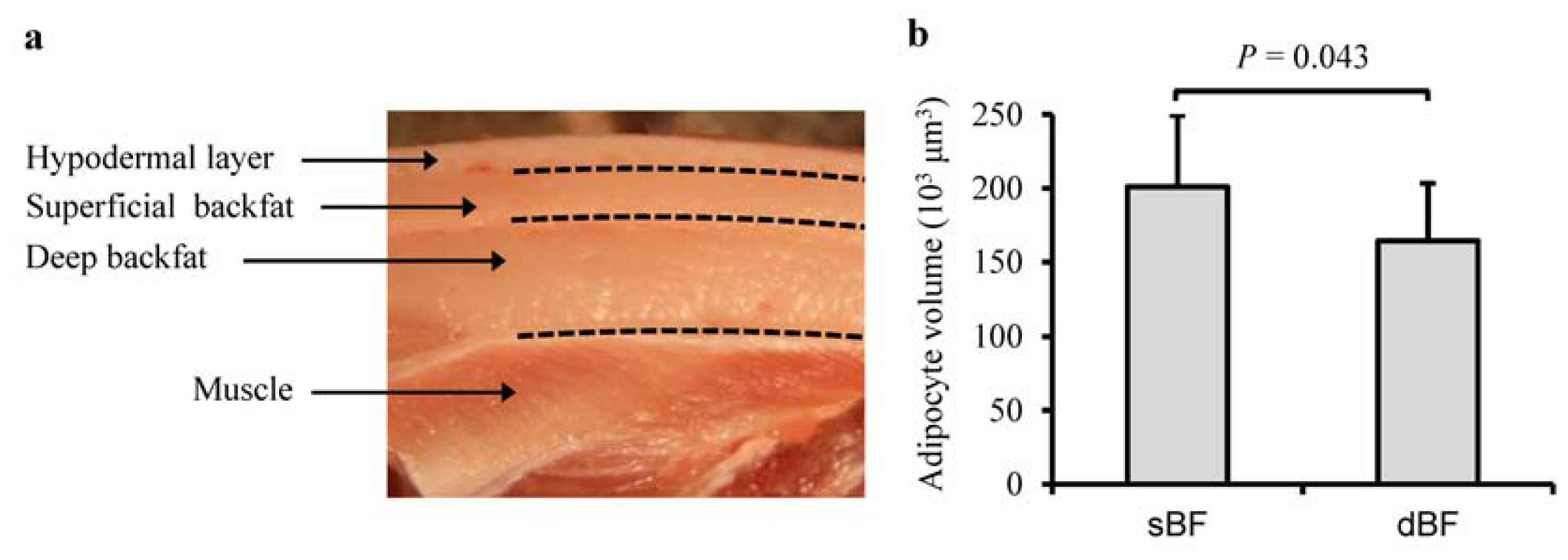
2.1. Characteristics of Porcine sBF and dBF Tissues
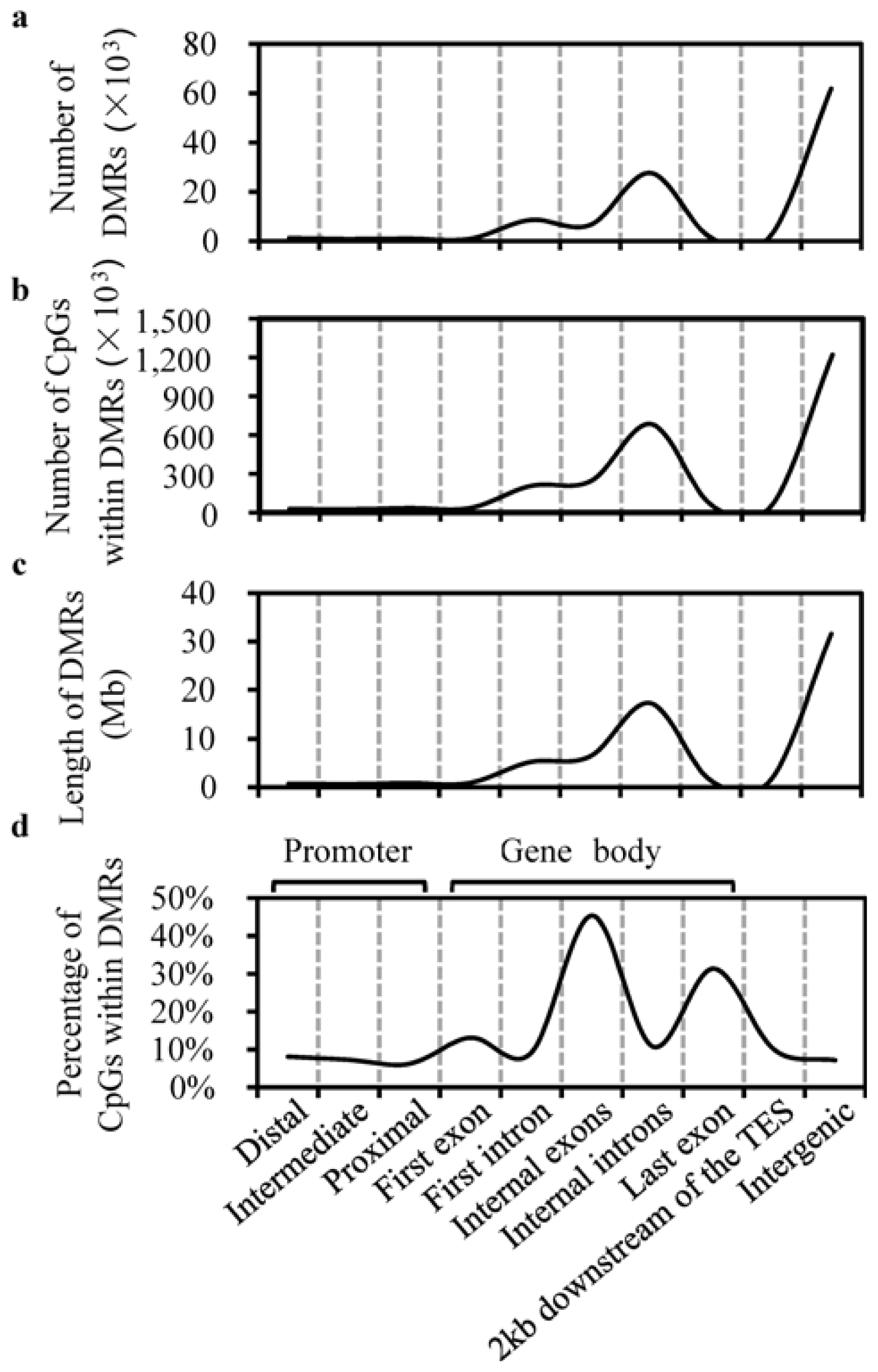
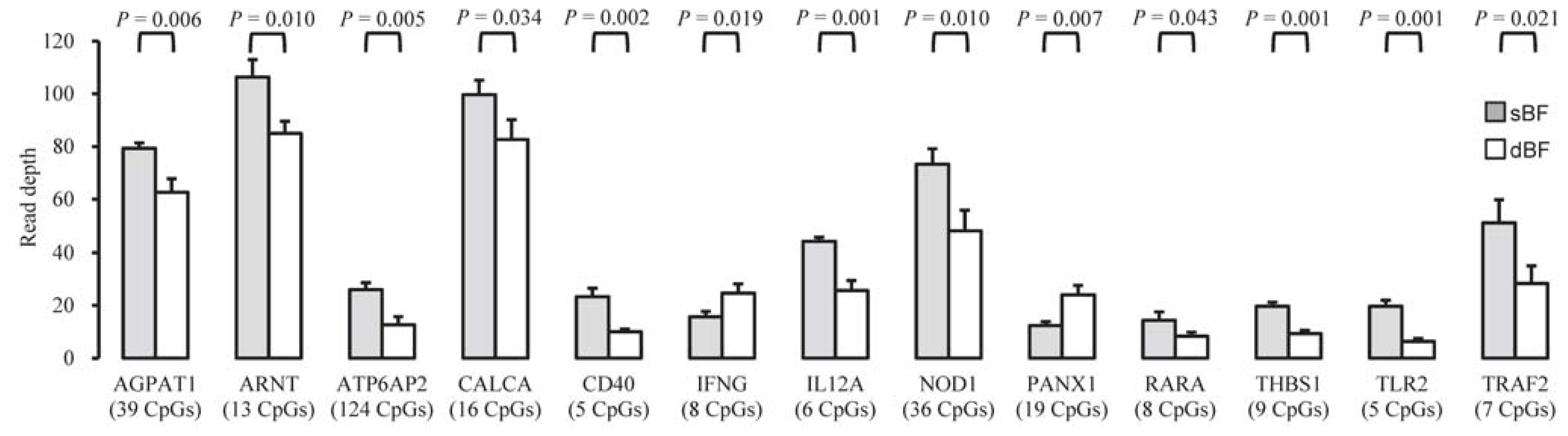
2.2. Characterization of DMRs
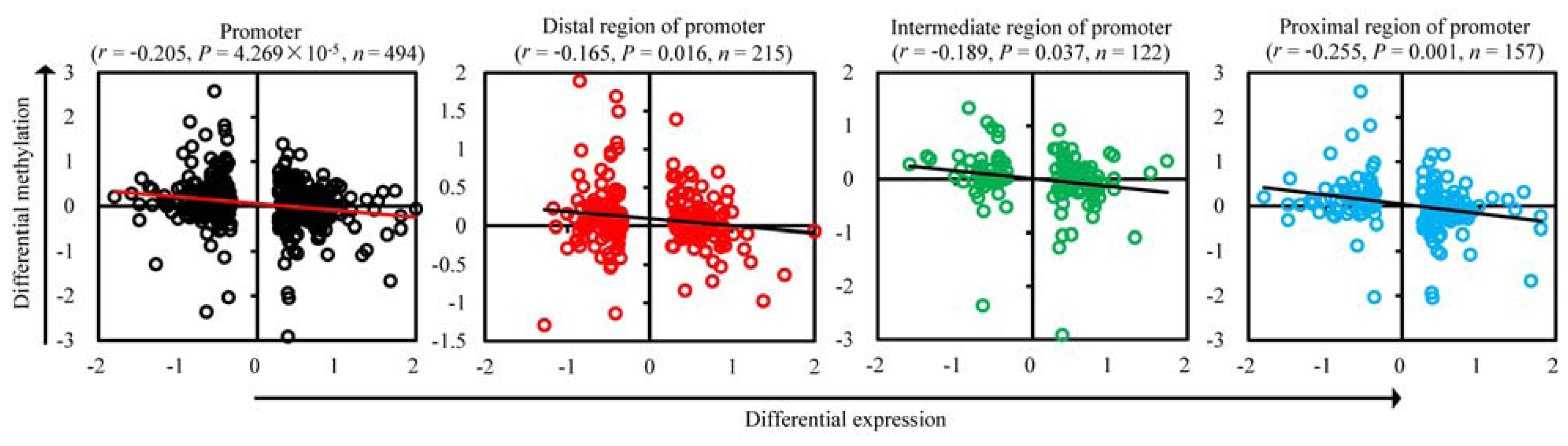
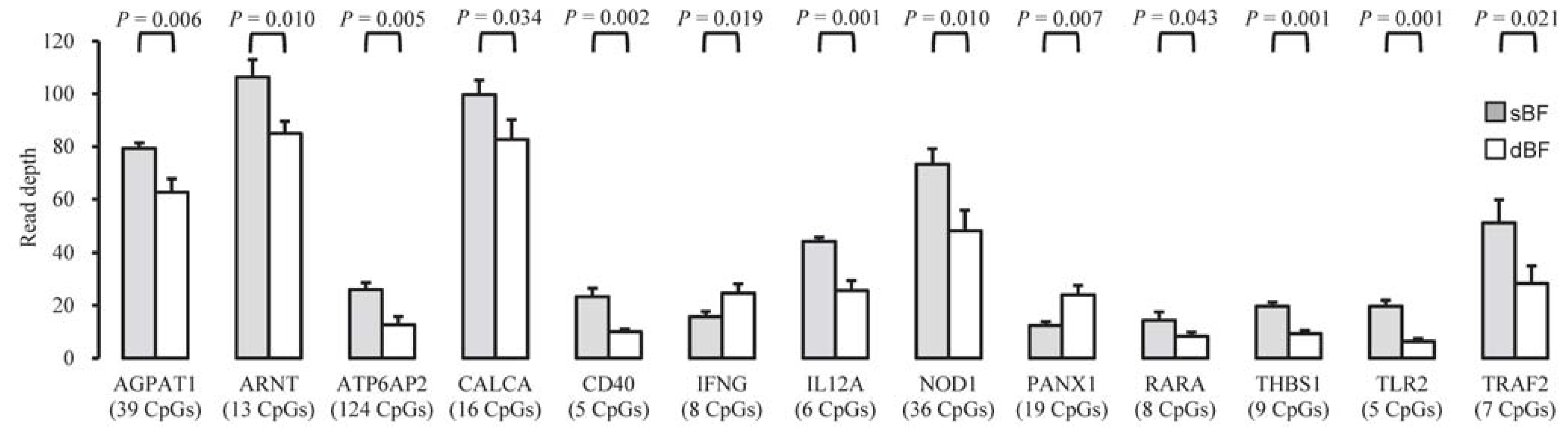
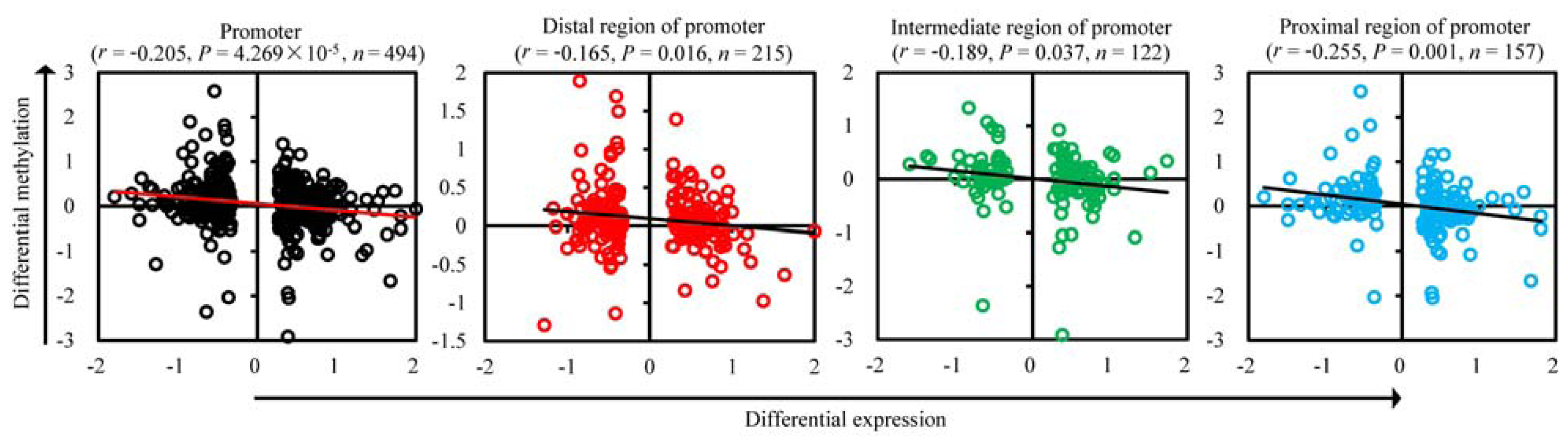
2.3. Correlation between mRNA Expression and Methylation in Promoters
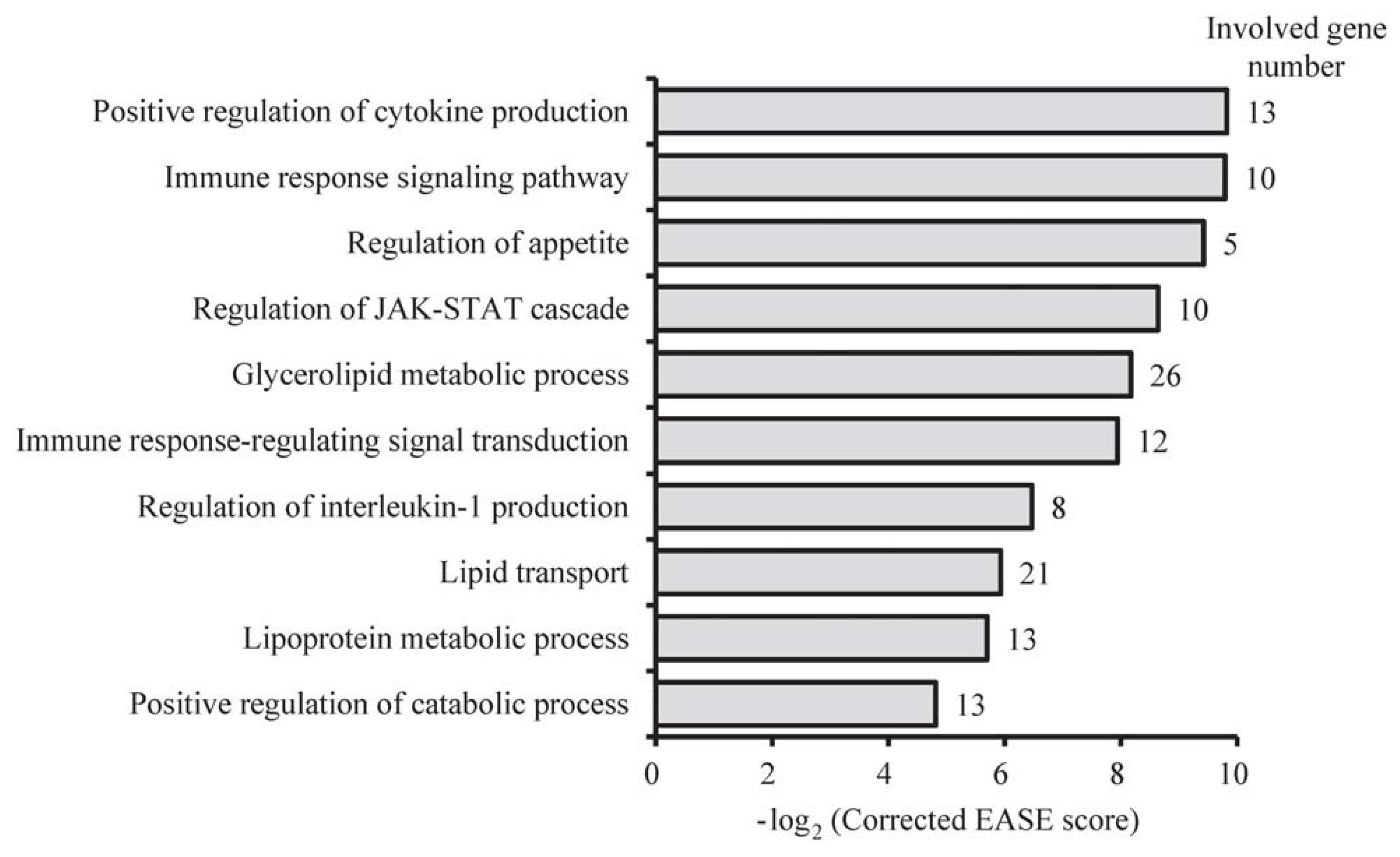
2.4. Functional Differences between sBF and dBF Tissues
3. Experimental Section
3.1. Animals and Tissue Collection
3.2. Measurement of Adipocyte Volume
3.3. Measurement of Fatty Acid Composition
3.4. MeDIP-Seq
3.5. Identification of DMRs
3.6. Definition of Genomic Elements
3.7. Gene Expression Microarray Analysis
4. Conclusions
Supplementary Information
ijms-13-07098-s001.pdfAcknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar]
- MacDougald, O.A.; Burant, C.F. The rapidly expanding family of adipokines. Cell Metab 2007, 6, 159–161. [Google Scholar]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev 2010, 11, 11–18. [Google Scholar]
- Wronska, A.; Kmiec, Z. Structural and biochemical characteristics of various white adipose tissue depots. Acta Physiol 2012, 205, 194–208. [Google Scholar]
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar]
- Yamamoto, Y.; Gesta, S.; Lee, K.Y.; Tran, T.T.; Saadatirad, P.; Kahn, C.R. Adipose depots possess unique developmental gene signatures. Obesity 2010, 18, 872–878. [Google Scholar]
- Sbarbati, A.; Accorsi, D.; Benati, D.; Marchetti, L.; Orsini, G.; Rigotti, G.; Panettiere, P. Subcutaneous adipose tissue classification. Eur. J. Histochem 2010, 54. [Google Scholar] [CrossRef]
- Walker, G.E.; Verti, B.; Marzullo, P.; Savia, G.; Mencarelli, M.; Zurleni, F.; Liuzzi, A.; di Blasio, A.M. Deep subcutaneous adipose tissue: A distinct abdominal adipose depot. Obesity 2007, 15, 1933–1943. [Google Scholar]
- Monzon, J.R.; Basile, R.; Heneghan, S.; Udupi, V.; Green, A. Lipolysis in adipocytes isolated from deep and superficial subcutaneous adipose tissue. Obes. Res 2002, 10, 266–269. [Google Scholar]
- Brook, C.G. Composition of human adipose tissue from deep and subcutaneous sites. Br. J. Nutr 1971, 25, 377–380. [Google Scholar]
- Spurlock, M.E.; Gabler, N.K. The development of porcine models of obesity and the metabolic syndrome. J. Nutr 2008, 138, 397–402. [Google Scholar]
- Rocha, D.; Plastow, G. Commercial pigs: An untapped resource for human obesity research? Drug Discov. Today 2006, 11, 475–477. [Google Scholar]
- Rankinen, T.; Zuberi, A.; Chagnon, Y.C.; Weisnagel, S.J.; Argyropoulos, G.; Walts, B.; Perusse, L.; Bouchard, C. The human obesity gene map: The 2005 update. Obesity 2006, 14, 529–644. [Google Scholar]
- Feinberg, A.P. Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nat. Biotechnol 2010, 28, 1049–1052. [Google Scholar]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet 2011, 12, 529–541. [Google Scholar]
- Garaulet, M.; Hernandez-Morante, J.J.; Lujan, J.; Tebar, F.J.; Zamora, S. Relationship between fat cell size and number and fatty acid composition in adipose tissue from different fat depots in overweight/obese humans. Int. J. Obes 2006, 30, 899–905. [Google Scholar]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol 2010, 28, 1057–1068. [Google Scholar]
- Koga, Y.; Pelizzola, M.; Cheng, E.; Krauthammer, M.; Sznol, M.; Ariyan, S.; Narayan, D.; Molinaro, A.M.; Halaban, R.; Weissman, S.M. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res 2009, 19, 1462–1470. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 2009, 4, 44–57. [Google Scholar]
- Iyer, A.; Fairlie, D.P.; Prins, J.B.; Hammock, B.D.; Brown, L. Inflammatory lipid mediators in adipocyte function and obesity. Nat. Rev. Endocrinol 2010, 6, 71–82. [Google Scholar]
- Schaffler, A.; Scholmerich, J. Innate immunity and adipose tissue biology. Trends Immunol 2010, 31, 228–235. [Google Scholar]
- Torres-Leal, F.L.; Fonseca-Alaniz, M.H.; Rogero, M.M.; Tirapegui, J. The role of inflamed adipose tissue in the insulin resistance. Cell Biochem. Funct 2010, 28, 623–631. [Google Scholar]
- Tran, T.T.; Yamamoto, Y.; Gesta, S.; Kahn, C.R. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab 2008, 7, 410–420. [Google Scholar]
- Amati, F.; Pennant, M.; Azuma, K.; Dube, J.J.; Toledo, F.G.; Rossi, A.P.; Kelley, D.E.; Goodpaster, B.H. Lower thigh subcutaneous and higher visceral abdominal adipose tissue content both contribute to insulin resistance. Obesity 2012, 5, 1115–1117. [Google Scholar]
- Satoor, S.N.; Puranik, A.S.; Kumar, S.; Williams, M.D.; Ghale, M.; Rahalkar, A.; Karandikar, M.S.; Shouche, Y.; Patole, M.; Bhonde, R. Location, location, location: Beneficial effects of autologous fat transplantation. Sci. Rep 2011, 1. [Google Scholar] [CrossRef]
- de Campos, R.; Hierro, E.; Ordóñez, J.; Bertol, T.; Terra, N.; de la Hoz, L. Fatty acid and volatile compounds from salami manufactured with yerba mate (IIex paraguariensis) extract and pork back fat and meat from pigs fed on diets with partial replacement of maize with rice bran. Food Chem 2007, 103, 1159–1167. [Google Scholar]
- Li, N.; Ye, M.; Li, Y.; Yan, Z.; Butcher, L.M.; Sun, J.; Han, X.; Chen, Q.; Zhang, X.; Wang, J. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods 2010, 52, 203–212. [Google Scholar]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty Acids | sBF | dBF | p Value |
|---|---|---|---|
| SFA | 35.63 ± 2.09 | 37.39 ± 2.19 | 0.005 |
| MUFA | 44.92 ± 0.80 | 43.54 ± 1.27 | 0.038 |
| PUFA | 19.45 ± 1.31 | 19.06 ± 0.97 | 0.235 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, M.; Wang, T.; Wu, H.; Zhang, J.; Zhou, C.; Jiang, A.; Li, R.; Li, X. Genome-Wide DNA Methylation Changes between the Superficial and Deep Backfat Tissues of the Pig. Int. J. Mol. Sci. 2012, 13, 7098-7108. https://doi.org/10.3390/ijms13067098
Li M, Wang T, Wu H, Zhang J, Zhou C, Jiang A, Li R, Li X. Genome-Wide DNA Methylation Changes between the Superficial and Deep Backfat Tissues of the Pig. International Journal of Molecular Sciences. 2012; 13(6):7098-7108. https://doi.org/10.3390/ijms13067098
Chicago/Turabian StyleLi, Mingzhou, Tao Wang, Honglong Wu, Jie Zhang, Chaowei Zhou, Anan Jiang, Ruiqiang Li, and Xuewei Li. 2012. "Genome-Wide DNA Methylation Changes between the Superficial and Deep Backfat Tissues of the Pig" International Journal of Molecular Sciences 13, no. 6: 7098-7108. https://doi.org/10.3390/ijms13067098
APA StyleLi, M., Wang, T., Wu, H., Zhang, J., Zhou, C., Jiang, A., Li, R., & Li, X. (2012). Genome-Wide DNA Methylation Changes between the Superficial and Deep Backfat Tissues of the Pig. International Journal of Molecular Sciences, 13(6), 7098-7108. https://doi.org/10.3390/ijms13067098