Mechanism of Cellular Oxidation Stress Induced by Asymmetric Dimethylarginine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
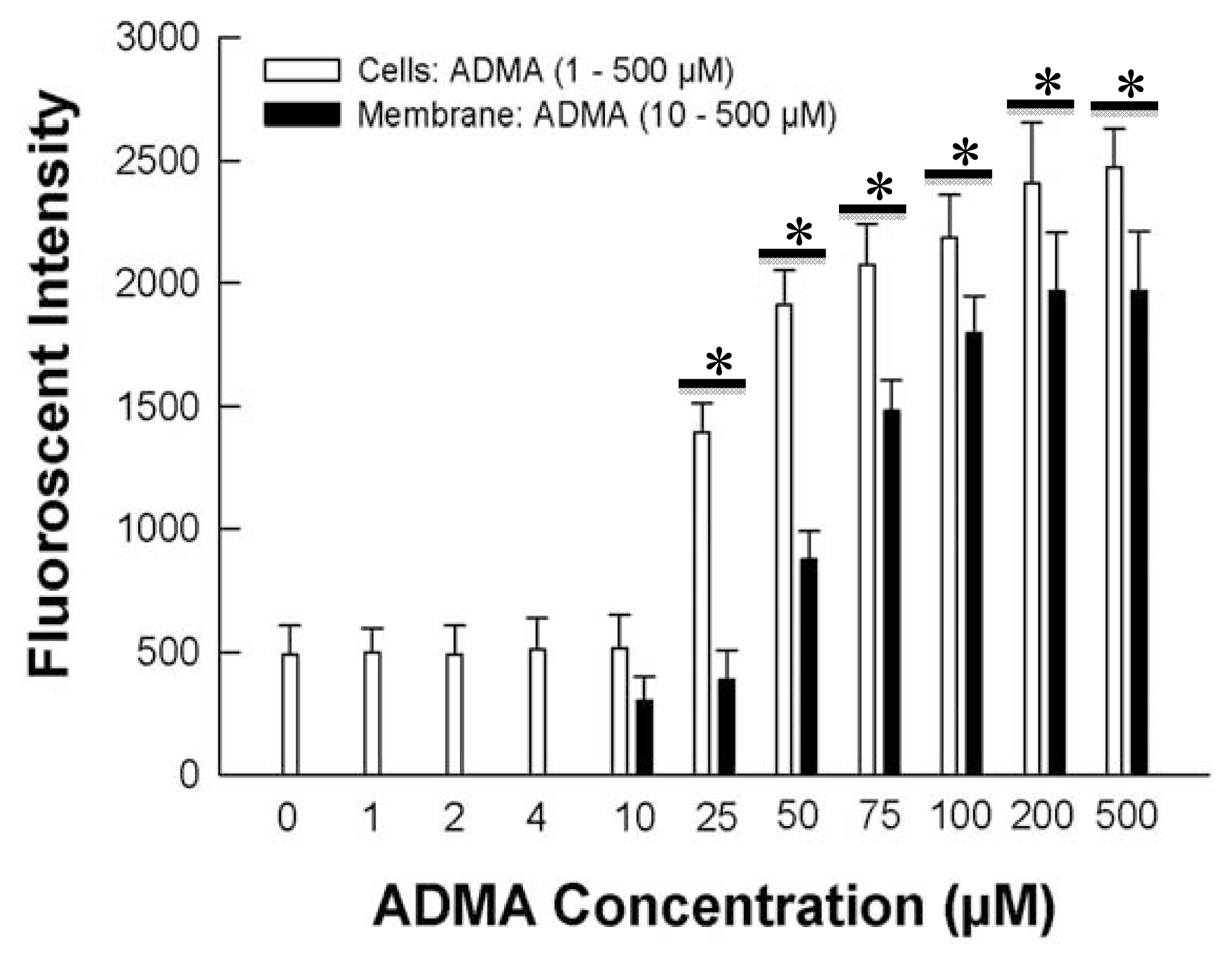
2. Results and Discussion
2.1. ADMA Induces Enchanced DHE Fluorescence in HUVEC Cells and Cell Membranes
2.2. Involvement of Membrane ADMA Transport and eNOS in Oxidative Stress
2.3. Effect of BH4 on ADMA-Induced Oxidative Stress
2.4. Nitric Oxide (NO) Bioavailability
2.5. Effects of Apocynin or Losartan on ADMA-Induced O2•− Production
3. Experimental Section
3.1. Supplies and Reagents
3.2. Cell Studies
3.3. Membrane Studies
3.4. DHE Fluorescence Measurement Using a Micro-Plate Reader
3.5. Inorganic Nitrite and Total Nitrite/Nitrate Determination
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
References
- MacAllister, R.J.; Fickling, S.A.; Whitley, G.S.; Vallance, P. Metabolism of methylarginines by human vasculature; implications for the regulation of nitric oxide synthesis. Br. J. Pharmacol 1994, 112, 43–48. [Google Scholar]
- McDermott, J.R. Studies on the catabolism of Ng-methylarginine, Ng, Ng-dimethylarginine and Ng, Ng-dimethylarginine in the rabbit. Biochem. J 1976, 154, 179–184. [Google Scholar]
- Rawal, N.; Rajpurohit, R.; Lischwe, M.A.; Williams, K.R.; Paik, W.K.; Kim, S. Structural specificity of substrate for S-adenosylmethionine:protein arginine N-methyltransferases. Biochim. Biophys. Acta 1995, 1248, 11–18. [Google Scholar]
- Juonala, M.; Viikari, J.S.; Alfthan, G.; Marniemi, J.; Kahonen, M.; Taittonen, L.; Laitinen, T.; Raitakari, O.T. Brachial artery flow-mediated dilation and asymmetrical dimethylarginine in the cardiovascular risk in young Finns study. Circulation 2007, 116, 1367–1373. [Google Scholar]
- Maas, R.; Schulze, F.; Baumert, J.; Lowel, H.; Hamraz, K.; Schwedhelm, E.; Koenig, W.; Boger, R.H. Asymmetric dimethylarginine, smoking, and risk of coronary heart disease in apparently healthy men: Prospective analysis from the population-based Monitoring of Trends and Determinants in Cardiovascular Disease/Kooperative Gesundheitsforschung in der Region Augsburg study and experimental data. Clin. Chem 2007, 53, 693–701. [Google Scholar]
- Schnabel, R.; Blankenberg, S.; Lubos, E.; Lackner, K.J.; Rupprecht, H.J.; Espinola-Klein, C.; Jachmann, N.; Post, F.; Peetz, D.; Bickel, C.; et al. Asymmetric dimethylarginine and the risk of cardiovascular events and death in patients with coronary artery disease: Results from the AtheroGene Study. Circ. Res 2005, 97, e53–e59. [Google Scholar]
- Surdacki, A.; Nowicki, M.; Sandmann, J.; Tsikas, D.; Boeger, R.H.; Bode-Boeger, S.M.; Kruszelnicka-Kwiatkowska, O.; Kokot, F.; Dubiel, J.S.; Froelich, J.C. Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J. Cardiovasc. Pharmacol 1999, 33, 652–658. [Google Scholar]
- Boger, R.H.; Bode-Boger, S.M.; Szuba, A.; Tsao, P.S.; Chan, J.R.; Tangphao, O.; Blaschke, T.F.; Cooke, J.P. Asymmetric dimethylarginine (ADMA): A novel risk factor for endothelial dysfunction: Its role in hypercholesterolemia. Circulation 1998, 98, 1842–1847. [Google Scholar]
- Sydow, K.; Schwedhelm, E.; Arakawa, N.; Bode-Boger, S.M.; Tsikas, D.; Hornig, B.; Frolich, J.C.; Boger, R.H. ADMA and oxidative stress are responsible for endothelial dysfunction in hyperhomocyst(e)inemia: Effects of L-arginine and B vitamins. Cardiovasc. Res 2003, 57, 244–252. [Google Scholar]
- Valkonen, V.P.; Paiva, H.; Salonen, J.T.; Lakka, T.A.; Lehtimaki, T.; Laakso, J.; Laaksonen, R. Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. Lancet 2001, 358, 2127–2128. [Google Scholar]
- Boger, R.H.; Bode-Boger, S.M.; Thiele, W.; Junker, W.; Alexander, K.; Frolich, J.C. Biochemical evidence for impaired nitric oxide synthesis in patients with peripheral arterial occlusive disease. Circulation 1997, 95, 2068–2074. [Google Scholar]
- Usui, M.; Matsuoka, H.; Miyazaki, H.; Ueda, S.; Okuda, S.; Imaizumi, T. Increased endogenous nitric oxide synthase inhibitor in patients with congestive heart failure. Life Sci 1998, 62, 2425–2430. [Google Scholar]
- Yoo, J.H.; Lee, S.C. Elevated levels of plasma homocyst(e)ine and asymmetric dimethylarginine in elderly patients with stroke. Atherosclerosis 2001, 158, 425–430. [Google Scholar]
- Gorenflo, M.; Zheng, C.; Werle, E.; Fiehn, W.; Ulmer, H.E. Plasma levels of asymmetrical dimethyl-L-arginine in patients with congenital heart disease and pulmonary hypertension. J. Cardiovasc. Pharmacol 2001, 37, 489–492. [Google Scholar]
- Kielstein, J.T.; Boger, R.H.; Bode-Boger, S.M.; Schaffer, J.; Barbey, M.; Koch, K.M.; Frolich, J.C. Asymmetric dimethylarginine plasma concentrations differ in patients with end-stage renal disease: Relationship to treatment method and atherosclerotic disease. J. Am. Soc. Nephrol 1999, 10, 594–600. [Google Scholar]
- Veresh, Z.; Racz, A.; Lotz, G.; Koller, A. ADMA impairs nitric oxide-mediated arteriolar function due to increased superoxide production by angiotensin II-NAD(P)H oxidase pathway. Hypertension 2008, 52, 960–966. [Google Scholar]
- Toth, J.; Racz, A.; Kaminski, P.M.; Wolin, M.S.; Bagi, Z.; Koller, A. Asymmetrical dimethylarginine inhibits shear stress-induced nitric oxide release and dilation and elicits superoxide-mediated increase in arteriolar tone. Hypertension 2007, 49, 563–568. [Google Scholar]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Antonopoulos, A.; Warrick, N.; van-Assche, T.; Cunnington, C.; Tousoulis, D.; Pillai, R.; Ratnatunga, C.; et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: Implications for endothelial function in human atherosclerosis. Eur. Heart J 2009, 30, 1142–1150. [Google Scholar]
- Hasegawa, K.; Wakino, S.; Tatematsu, S.; Yoshioka, K.; Homma, K.; Sugano, N.; Kimoto, M.; Hayashi, K.; Itoh, H. Role of asymmetric dimethylarginine in vascular injury in transgenic mice overexpressing dimethylarginie dimethylaminohydrolase 2. Circ. Res 2007, 101, e2–e10. [Google Scholar]
- Jiang, J.L.; Zhu, H.Q.; Chen, Z.; Xu, H.Y.; Li, Y.J. Angiotensin-converting enzyme inhibitors prevent LDL-induced endothelial dysfunction by reduction of asymmetric dimethylarginine level. Int. J. Cardiol 2005, 101, 153–155. [Google Scholar]
- Napoli, C.; Sica, V.; de Nigris, F.; Pignalosa, O.; Condorelli, M.; Ignarro, L.J.; Liguori, A. Sulfhydryl angiotensin-converting enzyme inhibition induces sustained reduction of systemic oxidative stress and improves the nitric oxide pathway in patients with essential hypertension. Am. Heart J 2004, 148. [Google Scholar] [CrossRef]
- Suda, O.; Tsutsui, M.; Morishita, T.; Tasaki, H.; Ueno, S.; Nakata, S.; Tsujimoto, T.; Toyohira, Y.; Hayashida, Y.; Sasaguri, Y.; et al. Asymmetric dimethylarginine produces vascular lesions in endothelial nitric oxide synthase-deficient mice: Involvement of renin-angiotensin system and oxidative stress. Arterioscler. Thromb. Vasc. Biol 2004, 24, 1682–1688. [Google Scholar]
- Antoniades, C.; Shirodaria, C.; Crabtree, M.; Rinze, R.; Alp, N.; Cunnington, C.; Diesch, J.; Tousoulis, D.; Stefanadis, C.; Leeson, P.; et al. Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation 2007, 116, 2851–2859. [Google Scholar]
- Zhao, H.; Kalivendi, S.; Zhang, H.; Joseph, J.; Nithipatikom, K.; Vasquez-Vivar, J.; Kalyanaraman, B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: Potential implications in intracellular fluorescence detection of superoxide. Free Radic. Biol. Med 2003, 34, 1359–1368. [Google Scholar]
- Mohan, S.; Wu, C.C.; Shin, S.; Fung, H.L. Continuous exposure to L-arginine induces oxidative stress and physiological tolerance in cultured human endothelial cells. Amino Acids 2011. [Google Scholar] [CrossRef]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res 2009, 60, 448–460. [Google Scholar]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar]
- Vasquez-Vivar, J.; Martasek, P.; Whitsett, J.; Joseph, J.; Kalyanaraman, B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: An EPR spin trapping study. Biochem. J 2002, 362, 733–739. [Google Scholar]
- Closs, E.I.; Graf, P.; Habermeier, A.; Cunningham, J.M.; Forstermann, U. Human cationic amino acid transporters hCAT-1, hCAT-2A, and hCAT-2B: Three related carriers with distinct transport properties. Biochemistry 1997, 36, 6462–6468. [Google Scholar]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem 1951, 193, 265–275. [Google Scholar]




© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mohan, S.; Fung, H.-L. Mechanism of Cellular Oxidation Stress Induced by Asymmetric Dimethylarginine. Int. J. Mol. Sci. 2012, 13, 7521-7531. https://doi.org/10.3390/ijms13067521
Mohan S, Fung H-L. Mechanism of Cellular Oxidation Stress Induced by Asymmetric Dimethylarginine. International Journal of Molecular Sciences. 2012; 13(6):7521-7531. https://doi.org/10.3390/ijms13067521
Chicago/Turabian StyleMohan, Srinidi, and Ho-Leung Fung. 2012. "Mechanism of Cellular Oxidation Stress Induced by Asymmetric Dimethylarginine" International Journal of Molecular Sciences 13, no. 6: 7521-7531. https://doi.org/10.3390/ijms13067521
APA StyleMohan, S., & Fung, H.-L. (2012). Mechanism of Cellular Oxidation Stress Induced by Asymmetric Dimethylarginine. International Journal of Molecular Sciences, 13(6), 7521-7531. https://doi.org/10.3390/ijms13067521