A DFT Study of Pyrrole-Isoxazole Derivatives as Chemosensors for Fluoride Anion

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
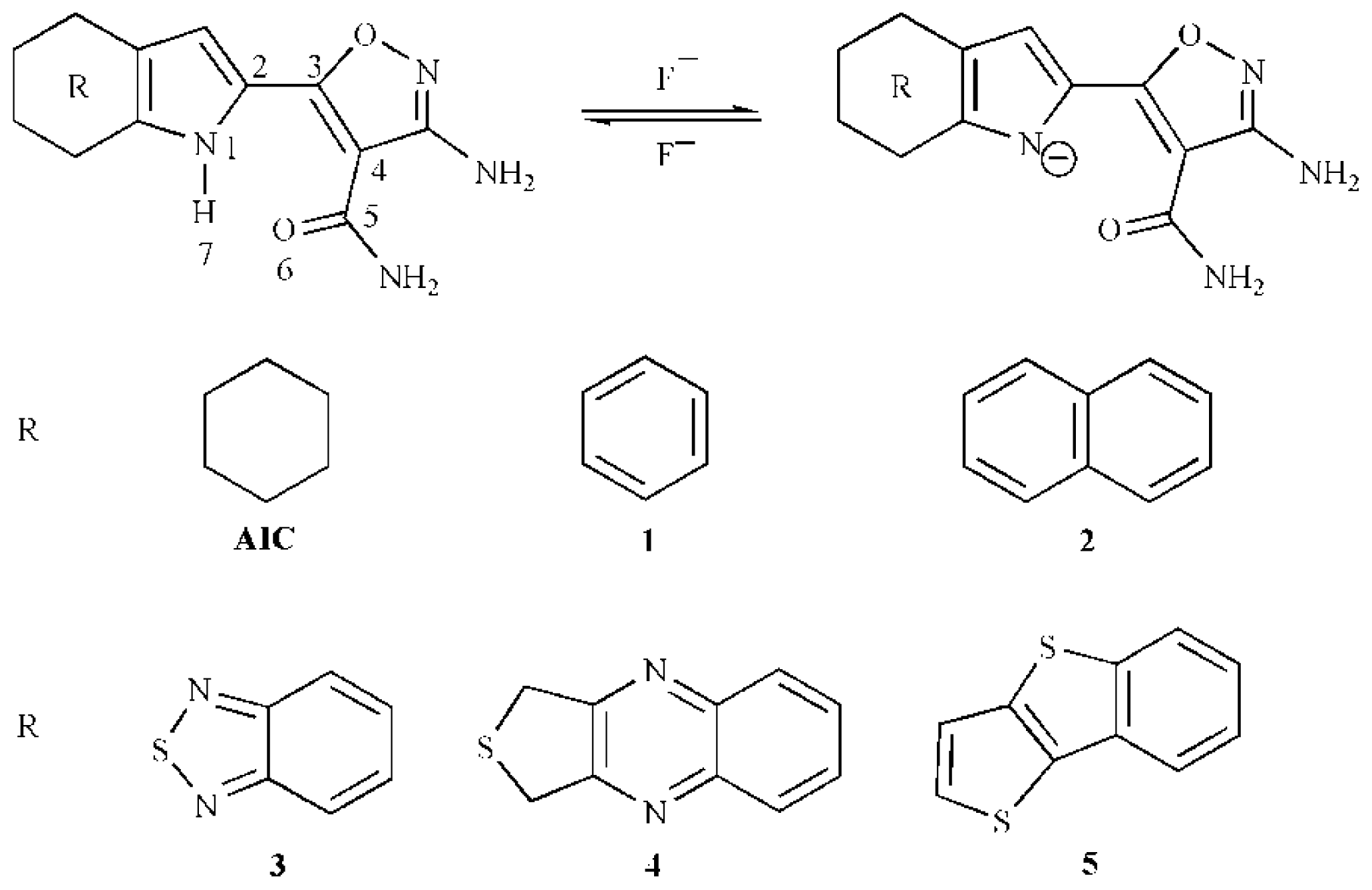
3.1. Host-Guest Interaction
3.2. AIM and NBO Analysis
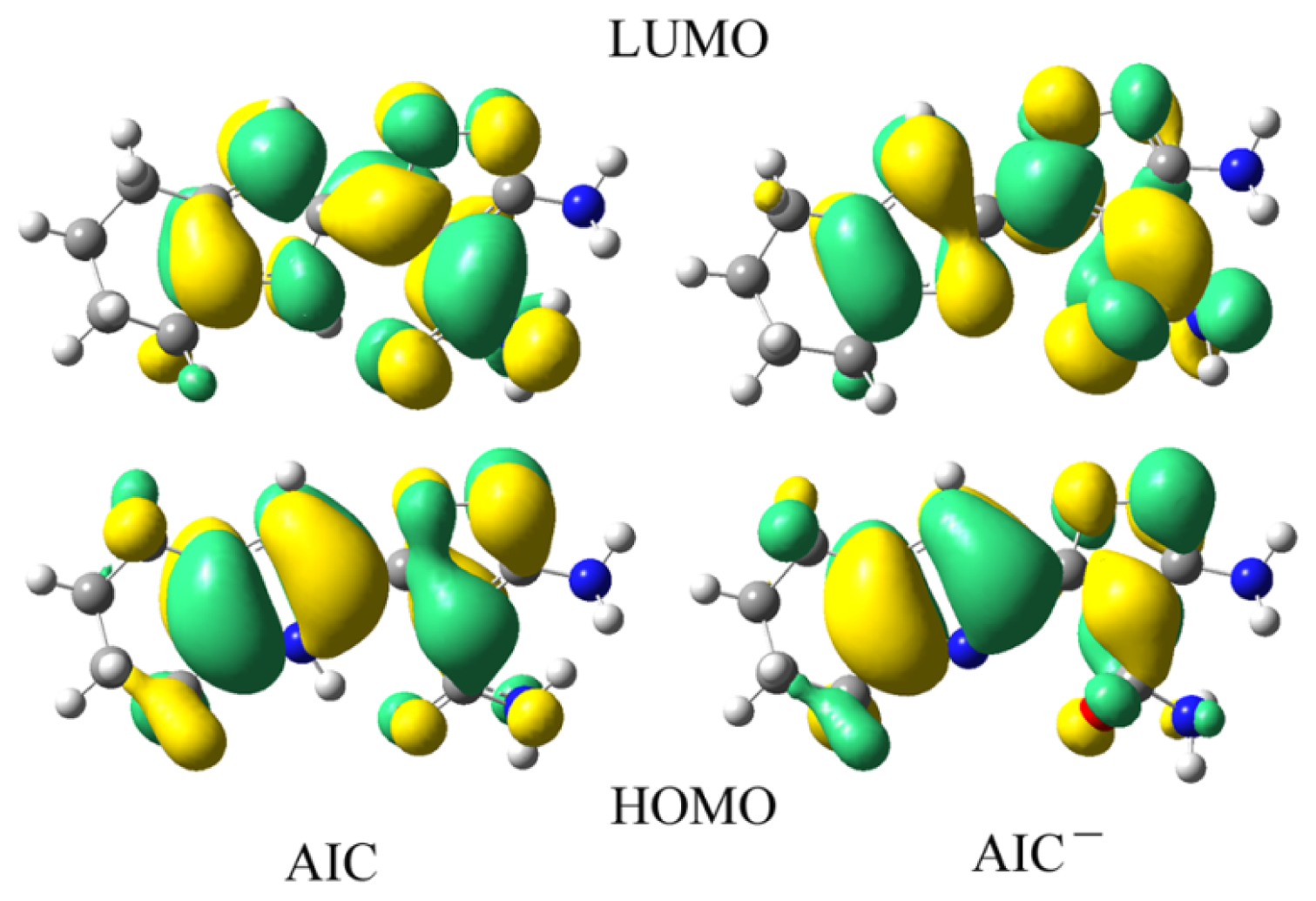
3.2. Electronic Properties
3.3. Optical Properties
4. Conclusions
Supplementary Materials
ijms-13-10986-s001.pdfAcknowledgments
References
- Bianchi, A.; Bowman-James, K.; Garcia-Espana, E. Supramolecular Chemistry of Anions; Wiley-VCH: New York, NY, USA; p. 1997.
- Deng, Y.; Chen, Y.; Cao, D.; Liu, Z.; Li, G. A cationic triarylborane as water-tolerant fluorescent chemosensor for fluoride anions. Sensors Actuator B-Chem 2010, 149, 165–169. [Google Scholar]
- Li, J.Q.; Li, X.Y. Multichannel photoinduced intramolecular electron-transfer excitations in a bis-naphthalimide spermine conjugate by time-dependent density functional theory. Phys. Chem. A 2007, 111, 13061–13068. [Google Scholar]
- Hagihara, S.; Tanaka, H.; Matile, S. Boronic acid converters for reactive hydrazide amplifiers: Polyphenol sensing in green tea with synthetic pores. J. Am. Chem. Soc 2008, 130, 5656–5657. [Google Scholar]
- Yoon, J.; Kim, S.K.; Singh, N.J.; Kim, K.S. Imidazolium receptors for the recognition of anions. Chem. Soc. Rev 2006, 35, 355–360. [Google Scholar]
- Gale, P.A. Structural and molecular recognition studies with acyclic anion receptors. Acc. Chem. Res 2006, 39, 465–475. [Google Scholar]
- Callan, J.F.; de Silva, A.P.; Magri, D.C. Luminescent sensors and switches in the early 21st century. Tetrahedron 2005, 61, 8551–8588. [Google Scholar]
- Martinez-Manez, R.; Sancanon, F. Fluorogenic and chromogenic chemosensors and reagents for anions. Chem. Rev 2003, 103, 4419–4476. [Google Scholar]
- Valeur, B. Molecular Fluorescence; Wiley-VCH: Weinhein, Germany; p. 2002.
- Coll, C.; Marínez-Máñez, R.; Dolores, M.M.; Sancenón, F.; Soto, J. A simple approach for the selective and sensitive colorimetric detection of anionic surfactants in water. Angew. Chem. Int. Ed 2007, 46, 1675–1678. [Google Scholar]
- Evans, L.S.; Gale, P.A.; Light, M.E.; Quesada, R. Anion binding vs deprotonation in colorimetric pyrrolylamidothiourea based anion sensors. Chem. Commun 2006, 965–967. [Google Scholar]
- Kirk, K.L. Biochemistry of the Halogens and Inorganic Halides; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Kleerekoper, M. The role of calcitonin in the prevention of osteoporosis. Endocrin. Metab. Clin. North. Am 1998, 27, 414–418. [Google Scholar]
- Zhang, S.W.; Swager, T.M. Fluorescent detection of chemical warfare agents: Functional group specific ratiometric chemosensors. J. Am. Chem. Soc 2003, 125, 3420–3421. [Google Scholar]
- Kim, S.K.; Yoon, J. A new fluorescent PET chemosensor for fluoride ions. Chem. Commun 2002, 770–771. [Google Scholar]
- Peng, X.; Wu, Y.; Fan, J.; Tian, M.; Han, K. Colorimetric and ratiometric fluorescence sensing of fluoride: Tuning selectivity in proton transfer. J. Org. Chem 2005, 70, 10524–10531. [Google Scholar]
- Li, Z.; Zhang, J. An efficient theoretical study on host–guest interactions of a fluoride chemosensor with F−, Cl−, and Br−. Chem. Phys 2006, 331, 159–163. [Google Scholar]
- Thiagarajan, V.; Ramamurthy, P. Specific optical signalling of anions via intramolecular charge transfer pathway based on acridinedione fluorophore. J. Lumin 2007, 126, 886–892. [Google Scholar]
- Kim, S.K.; Bok, J.H.; Bartsch, R.A.; Lee, J.Y.; Kim, J.S. A fluoride-selective PCT chemosensor based on formation of a static pyrene excimer. Org. Lett 2005, 7, 4839–4842. [Google Scholar]
- Chow, C.F.; Chiu, B.K.W.; Lam, M.H.W.; Wong, W.Y. A trinuclear heterobimetallic Ru(II)/Pt(II) complex as a chemodosimeter selective for sulfhydryl-containing amino acids and peptides. J. Am. Chem. Soc 2003, 125, 7802–7803. [Google Scholar]
- Yang, Z.; Zhang, K.; Gong, F.; Li, S.; Chen, J.; Ma, J.S.; Sobenina, L.N.; Mikhaleva, A.I.; Yang, G.; Trofimov, B.A. A new fluorescent chemosensor for fluoride anion based on a pyrrole–isoxazole derivative. Beilstein. J. Org. Chem 2011, 7, 46–52. [Google Scholar]
- Frisch, M.J.T.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc: Wallingford, CT, USA; p. 2009.
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys 1996, 105, 11024–11031. [Google Scholar]
- Handy, N.C. The molecular physics lecture 2004: (i) Density functional theory; (ii) Quantum Monte Carlo. Mol. Phys 2004, 102, 2399–2499. [Google Scholar]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys 2004, 120, 8425–8433. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett 2004, 393, 51–57. [Google Scholar]
- Kobayashi, R.; Amos, R.D. The application of CAM-B3LYP to the charge-transfer band problem of the zincbacteriochlorin–bacteriochlorin complex. Chem. Phys. Lett 2006, 420, 106–109. [Google Scholar]
- Kerkines, I.S.K.; Petsalakis, I.D.; Theodorakopoulos, G. Excited-state intramolecular proton transfer in hydroxyoxime-based chemical sensors. J. Phys. Chem. A 2011, 115, 834–840. [Google Scholar]
- Rostov, I.V.; Amos, R.D.; Kobayashi, R.; Scalmani, G.; Frisch, M.J. Studies of the ground and excited-state surfaces of the retinal chromophore using CAM-B3LYP. J. Phys. Chem. B 2010, 114, 5547–5555. [Google Scholar]
- Liu, X.; Zhang, X.; Hou, Y.; Teng, F.; Lou, Z. Theoretical investigation on properties of the ground and lowest excited states of a red emitter with donor-p-acceptor structure. Chem. Phys 2011, 381, 100–104. [Google Scholar]
- Monari, A.; Assfeld, X.; Beley, M.; Gros, P.C. Theoretical study of new ruthenium-based dyes for dye-sensitized solar cells. J. Phys. Chem. A 2011, 115, 3596–3603. [Google Scholar]
- Cornard, J.P.; Lapouge, C. Absorption spectra of caffeic acid, caffeate and their 1:1 complex with Al(III): Density functional theory and time-dependent density functional theory investigations. J. Phys. Chem. A 2006, 110, 7159–7166. [Google Scholar]
- Bader, R.F.W. A bond path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar]
- Biegler-König, F. AIM2000; University of Applied Sciences Bielefeld: Bielefeld, Germany, 2000. [Google Scholar]
- Hobza, P.; Havlas, Z. Blue-shifting hydrogen bonds. Chem. Rev 2000, 100, 4253–4264. [Google Scholar]
- Steiner, T.; Desiraju, G.R. Distinction between the weak hydrogen bond and the van der waals interaction. Chem. Commun 1998, 891–892. [Google Scholar]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed 2002, 41, 48–76. [Google Scholar]
- Amendola, V.; Esteban-Gómez, D.; Fabbrizzi, L.; Licchelli, M. What anions do to N–H-containing receptors. Acc. Chem. Res 2006, 39, 343–353. [Google Scholar]
- Schwenke, D.W.; Truhlar, D.G. Systematic study of basis set superposition errors in the calculated interaction energy of two HF molecules. J. Chem. Phys 1985, 82, 2418–2426. [Google Scholar]
- Sidorkin, V.F.; Doronina, E.P.; Chipanina, N.N.; Aksamentova, T.N.; Shainyan, B.A. Bifurcate hydrogen bonds. Interaction of intramolecularly H-bonded systems with Lewis bases. J. Phys. Chem. A 2008, 112, 6227–6234. [Google Scholar]
- Nagaraju, M.; Narahari, S.G. Effect of alkyl substitution on H-bond strength of substituted amide-alcohol complexes. J. Mol. Model 2011, 17, 1801–1816. [Google Scholar]
- Karabıyık, H.; Sevinçek, R.; Petek, H.; Aygün, M. Aromaticity balance, π-electron cooperativity and H-bonding properties in tautomerism of salicylideneaniline: The quantum theory of atoms in molecules (QTAIM) approach. J. Mol. Model 2011, 7, 1295–1309. [Google Scholar]
- Abramov, Y.A. On the possibility of kinetic energy density evaluation from the experimental electron-density distribution. Acta. Crystallogr. A 1997, 53, 264–272. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK; p. 1994.
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett 1998, 285, 170–173. [Google Scholar]
- Coppens, P.; Abramov, Y.; Carducci, M.; Korjov, B.; Novozhilova, I.; Alhambra, C.; Pressprich, M.R. Experimental charge densities and intermolecular interactions: Electrostatic and topological analysis of dl-histidine. J. Am. Chem. Soc 1999, 121, 2585–2593. [Google Scholar]
- Musin, R.N.; Mariam, Y.H. An integrated approach to the study of intramolecular hydrogen bonds in malonaldehyde enol derivatives and naphthazarin: Trend in energetic versus geometrical consequences. J. Phys. Org. Chem 2006, 19, 425–444. [Google Scholar]
- Nakanishi, W.; Hayashi, S.; Narahara, K. Atoms-in-molecules dual parameter analysis of weak to strong interactions: Behaviors of electronic energy densities versus laplacian of electron densities at bond critical points. J. Phys. Chem. A 2008, 112, 13593–13599. [Google Scholar]
- Forés, M.; Duran, M.; Solà, M.; Adamowicz, L. Excited-state intramolecular proton transfer and rotamerism of 2-(2′-hydroxyvinyl)benzimidazole and 2-(2′-hydroxyphenyl)imidazole. J. Phys. Chem. A 1999, 103, 4413–4420. [Google Scholar]
- Helal, A.; Rashid, M.H.O.; Choi, C.-H.; Kim, H.-S. Chromogenic and fluorogenic sensing of Cu2+ based on coumarin. Tetrahedron 2011, 67, 2794–2802. [Google Scholar]



{kind=link}
{kind=link}
| without BSSE | with BSSE | |||||
|---|---|---|---|---|---|---|
| Complexes | RH7···N1 | RH7–X | θN1···H7–X | RH7···N1 | RH7–X | θN1···H7–X |
| AIC | 1.025 | |||||
| AIC−·HF | 1.467 | 1.031 | 170.8 | 1.472 | 1.030 | 170.8 |
| AIC·Cl− | 1.046 | 2.126 | 156.6 | 1.045 | 2.139 | 156.1 |
| AIC·Br− | 1.036 | 2.320 | 153.2 | 1.032 | 2.479 | 147.4 |
| AIC·AcO− | 1.069 | 1.614 | 170.7 | 1.067 | 1.630 | 169.5 |
| AIC·H2PO4− | 1.049 | 1.671 | 176.1 | 1.048 | 1.686 | 175.3 |
| n | n−·HF | n·Cl− | n·Br− | n·AcO− | n·H2PO4− |
|---|---|---|---|---|---|
| AIC | −30.7 | −6.9 | −4.1 | −12.0 | −16.4 |
| 1 | −34.9 | −9.4 | −6.3 | −14.6 | −13.5 |
| 2 | −37.4 | −11.2 | −8.0 | −16.4 | −14.7 |
| 3 | −41.9 | −14.3 | −7.3 | −19.1 | −23.6 |
| 4 | −42.2 | −14.2 | −6.9 | −18.4 | −16.5 |
| 5 | −39.1 | −11.9 | −8.6 | −17.9 | −23.4 |
| H7···N1 | H7–X | |||||
|---|---|---|---|---|---|---|
| X | ρ(r)bcp | ∇2ρ(r)bcp | EHB | ρ(r)bcp | ∇2ρ(r)bcp | EHB |
| F | 0.0926 | 0.0429 | −29.6 | 0.2506 | −1.026 | −145.9 |
| Cl | 0.3080 | −1.6563 | −156.8 | 0.0302 | 0.0609 | −6.1 |
| Br | 0.3206 | −1.7499 | −163.1 | 0.0183 | 0.040968 | −3.1 |
| AcO | 0.2887 | −1.5106 | −146.7 | 0.0545 | 0.138816 | −13.1 |
| H2PO4 | 0.3057 | −1.6347 | −155.9 | 0.0459 | 0.131748 | −10.9 |
| Neutral | Anion | |||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | λabs | f | Assignments | Exp * | λabs | f | Assignments | Exp * |
| AIC | 341 | 0.67 | H→L (0.70) | 340 | 375 | 0.55 | H→L (0.70) | 375 |
| 1 | 334 | 0.45 | H-1→L (0.69) | 346 | 0.64 | H-1→L (0.69) | ||
| 2 | 357 | 0.80 | H-1→L (0.69) | 366 | 0.80 | H-1→L (0.68) | ||
| 3 | 368 | 0.53 | H-1→L (0.62) | 388 | 0.80 | H-1→L (0.66) H→L+1 (0.23) | ||
| 4 | 689 | 0.07 | H→L (0.71) | 457 | 0.21 | H-1→L (0.54) H-2→L (0.38) | ||
| 5 | 379 | 0.93 | H-1→L (0.69) | 383 | 0.93 | H-1→L (0.69) H→L + 1 (0.13) | ||
| neutral | anion | |||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | λfl | f | Assignments | Exp* | λfl | f | Assignments | Exp* |
| AIC | 409 | 0.42 | H←L (0.68) H-1←L (0.10) | 400 | 465 | 0.02 | H←L + 1 (0.75) | 432 |
| 1 | 471 | 0.27 | H←L (0.68) H-1←L (0.16) | 409 | 0.48 | H-1←L (0.69) | ||
| 2 | 413 | 0.65 | H←L (0.12) H-1←L (0.65) | 721 | 0.04 | H←L (0.70) | ||
| 3 | 416 | 0.37 | H←L + 1 (0.28) H-1←L (0.48) | 527 | 0.07 | H←L + 1 (0.69) | ||
| 4 | 579 | 0.02 | H←L + 1 (0.45) H-1←L (0.53) | 514 | 0.25 | H←L + 1 (0.29) H-1←L (0.60) | ||
| 5 | 462 | 0.63 | H-1←L (0.66) H←L (0.13) | 438 | 0.85 | H←L + 1 (0.36) H-1←L (0.59) | ||
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jin, R.; Sun, W.; Tang, S. A DFT Study of Pyrrole-Isoxazole Derivatives as Chemosensors for Fluoride Anion. Int. J. Mol. Sci. 2012, 13, 10986-10999. https://doi.org/10.3390/ijms130910986
Jin R, Sun W, Tang S. A DFT Study of Pyrrole-Isoxazole Derivatives as Chemosensors for Fluoride Anion. International Journal of Molecular Sciences. 2012; 13(9):10986-10999. https://doi.org/10.3390/ijms130910986
Chicago/Turabian StyleJin, Ruifa, Weidong Sun, and Shanshan Tang. 2012. "A DFT Study of Pyrrole-Isoxazole Derivatives as Chemosensors for Fluoride Anion" International Journal of Molecular Sciences 13, no. 9: 10986-10999. https://doi.org/10.3390/ijms130910986
APA StyleJin, R., Sun, W., & Tang, S. (2012). A DFT Study of Pyrrole-Isoxazole Derivatives as Chemosensors for Fluoride Anion. International Journal of Molecular Sciences, 13(9), 10986-10999. https://doi.org/10.3390/ijms130910986