Differences in the Structure of the Gut Bacteria Communities in Development Stages of the Chinese White Pine Beetle (Dendroctonus armandi)



Abstract
:1. Introduction
2. Results and Discussion
2.1. Bacterial Diversity Analysis
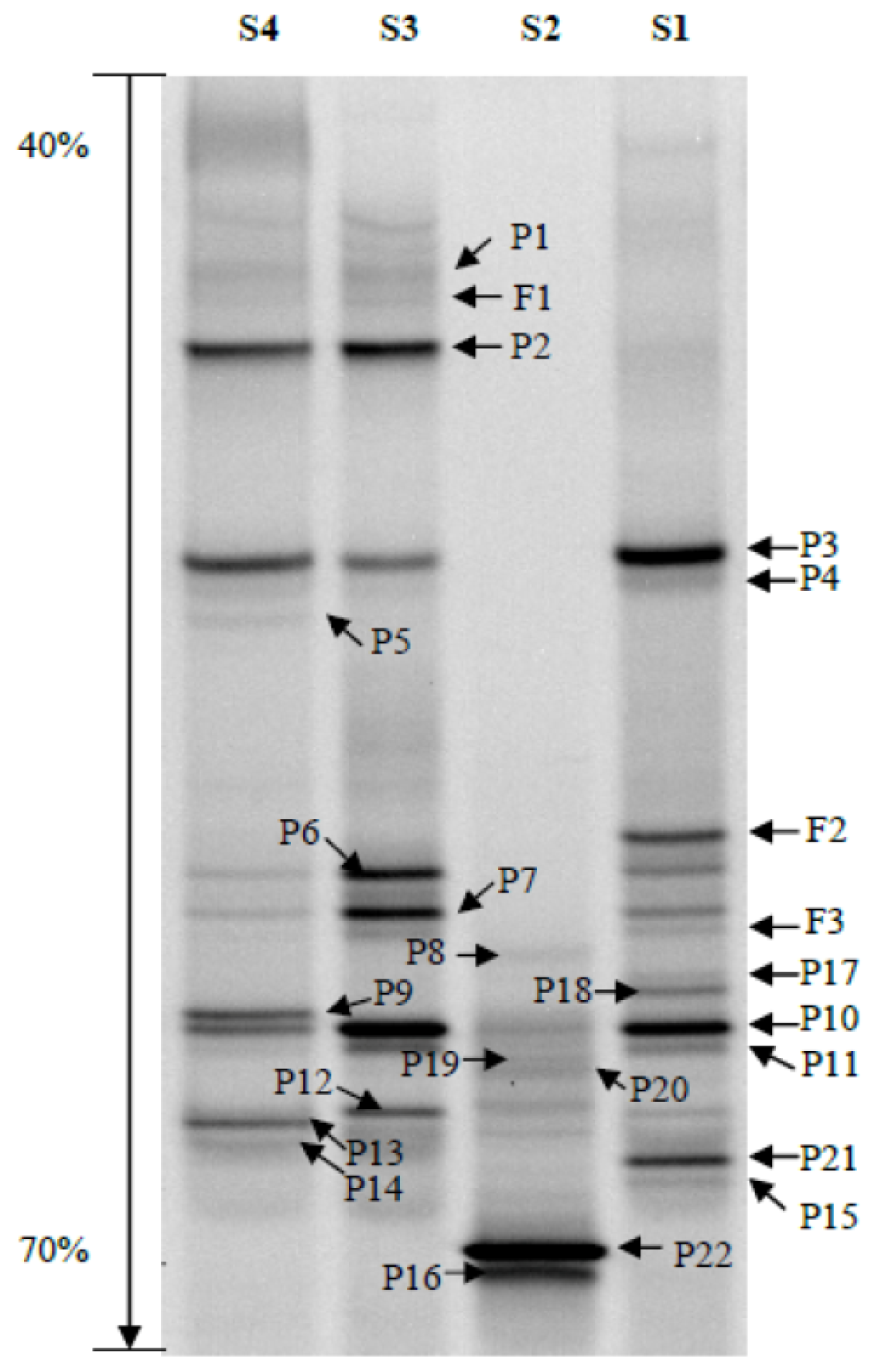
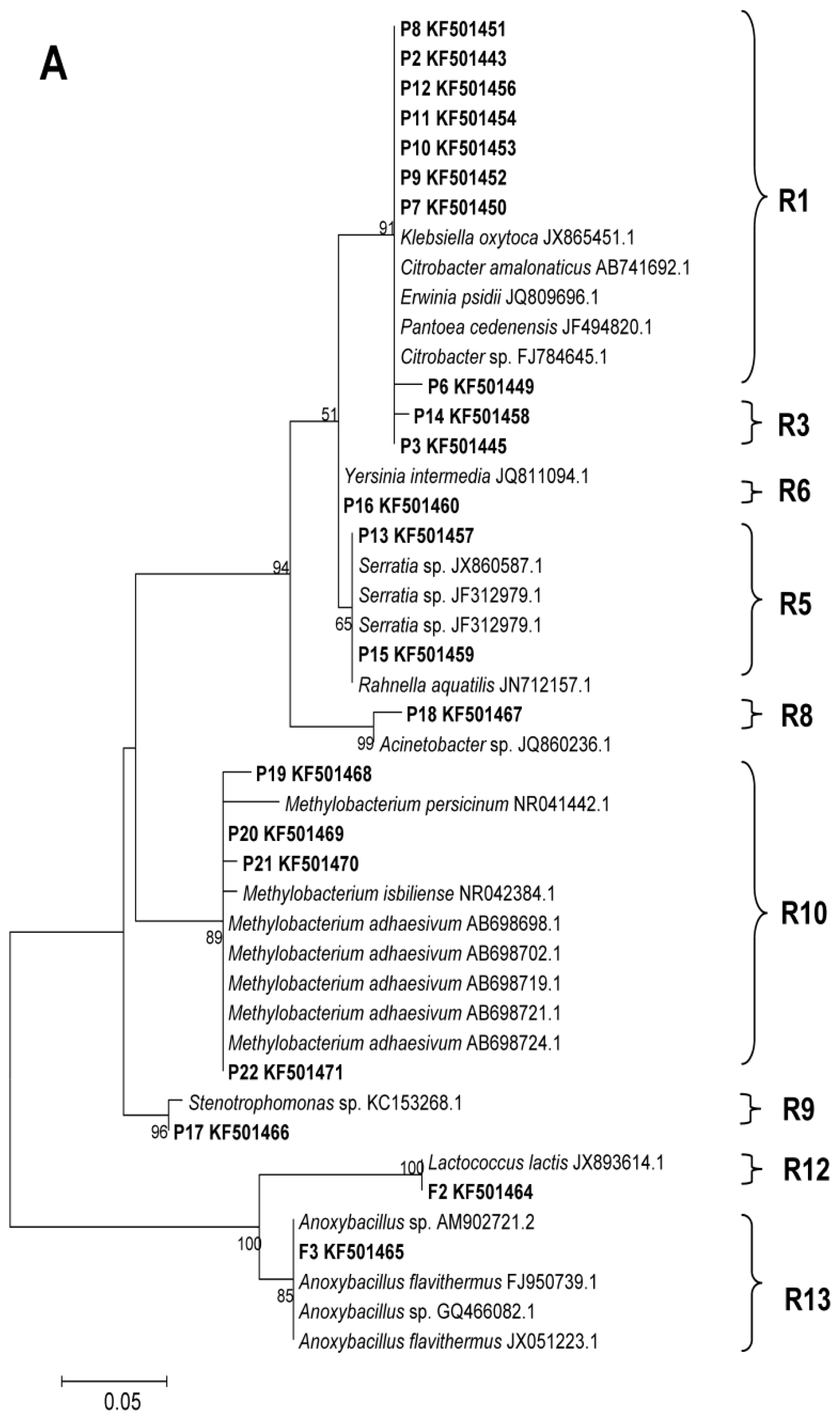
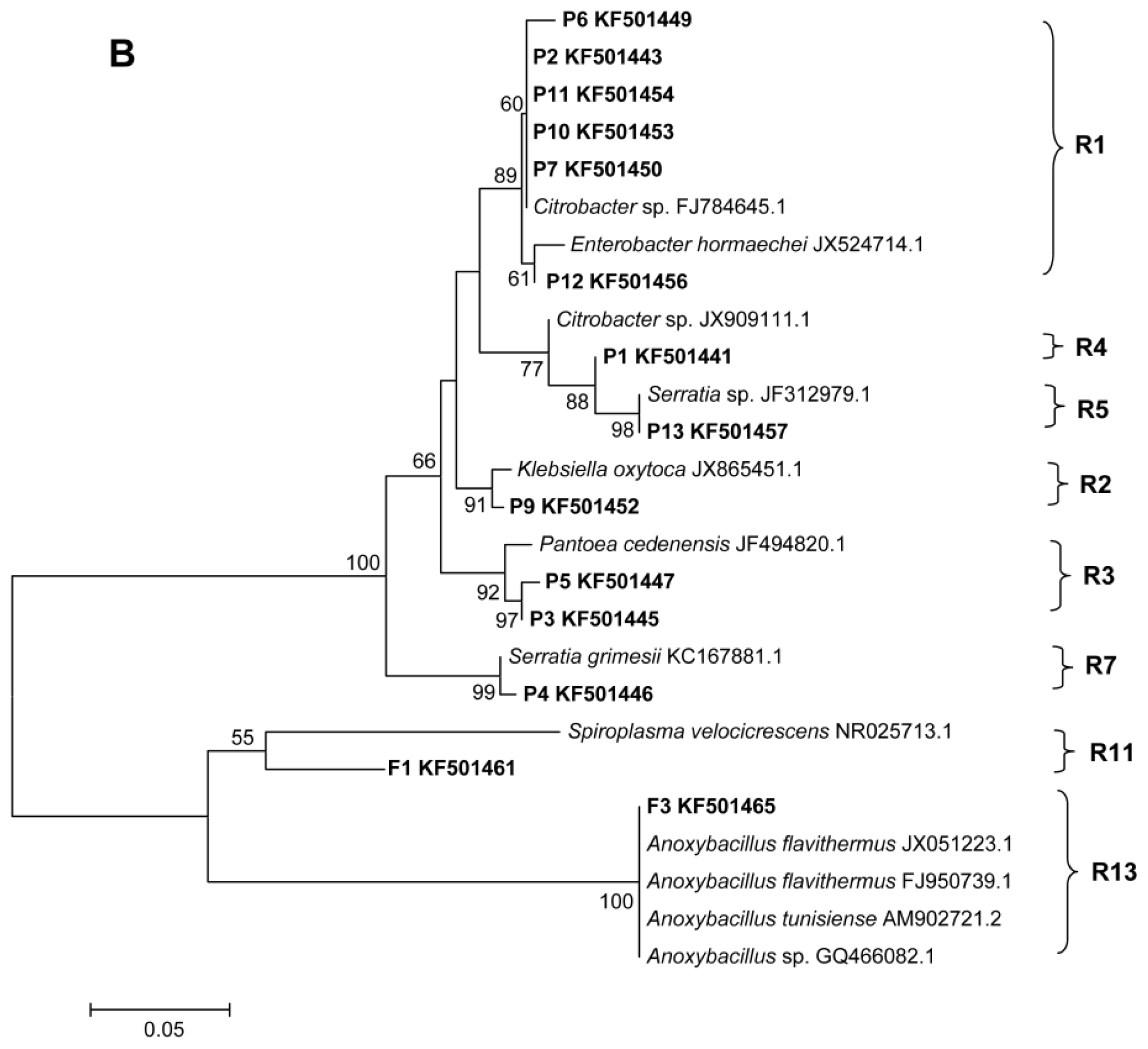
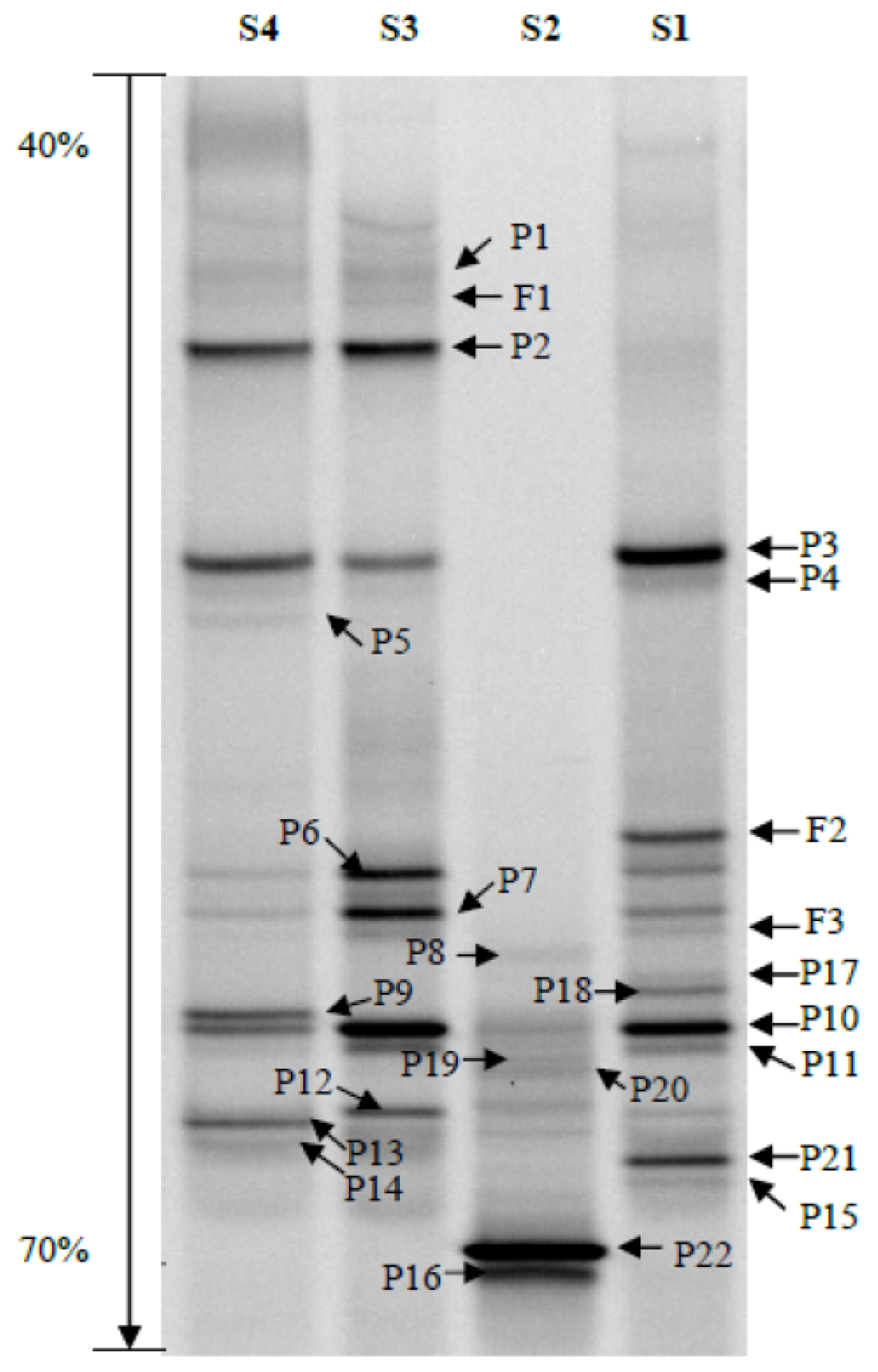
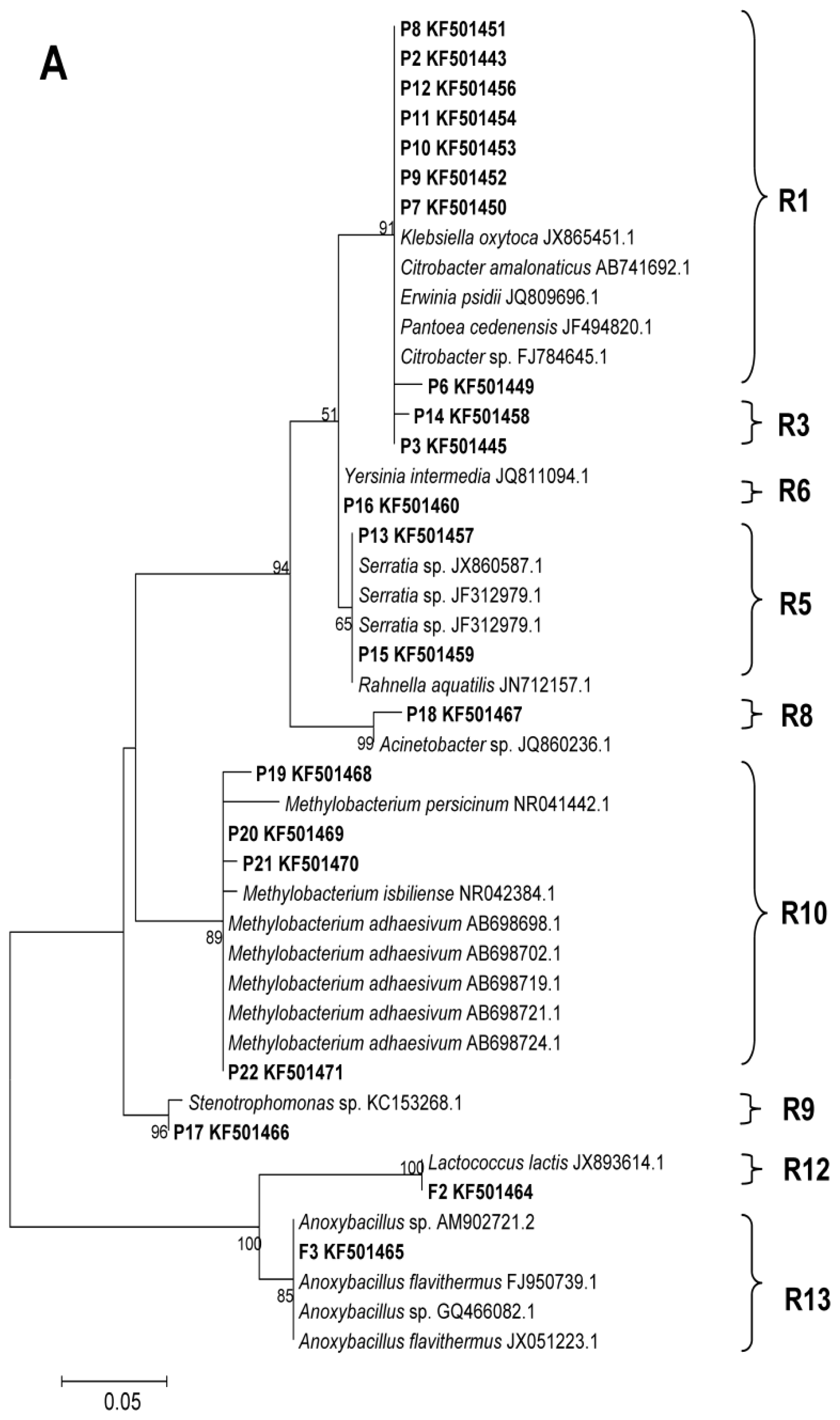
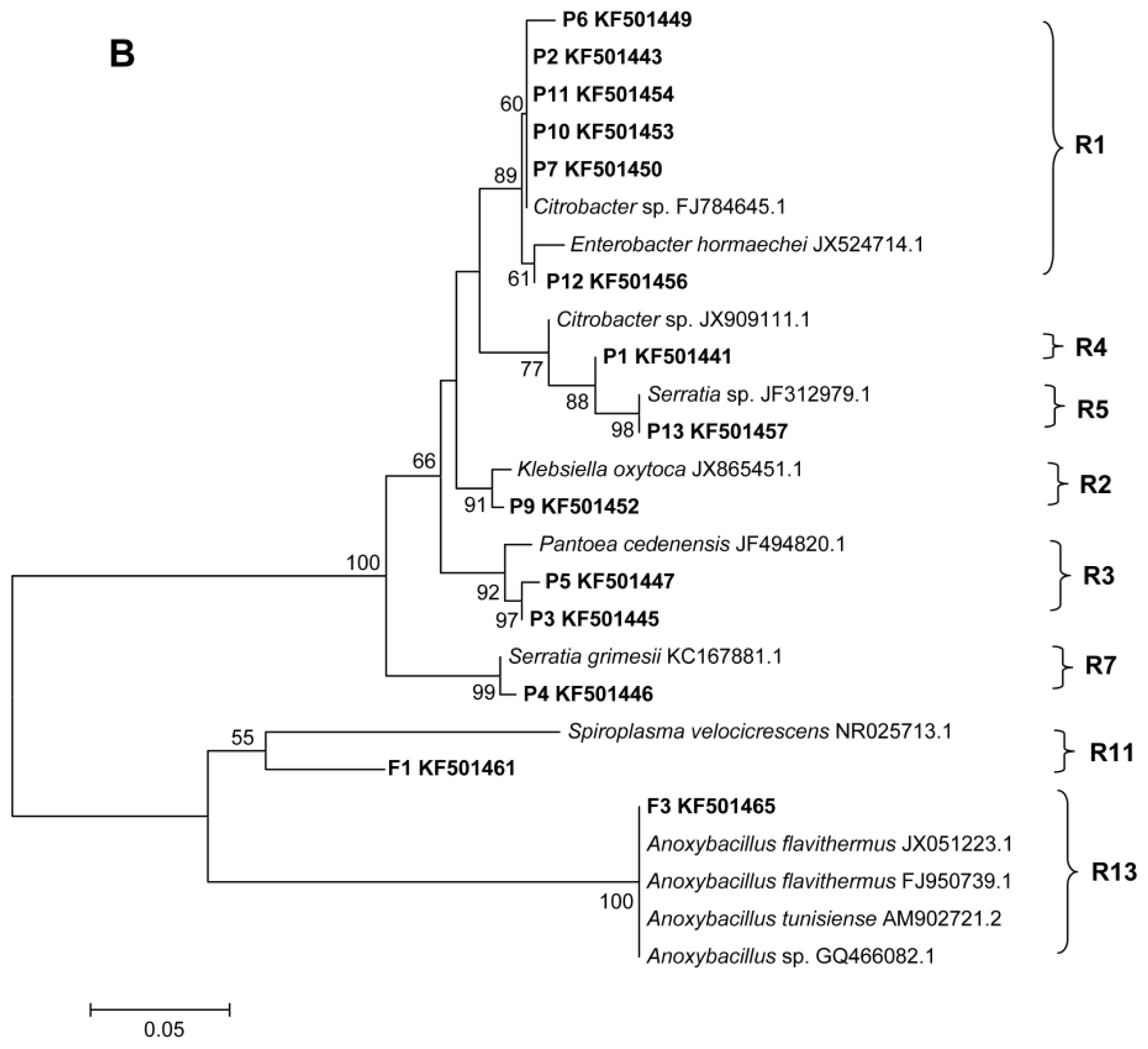
2.2. Phylogenetic Analyses and Dominant Taxa
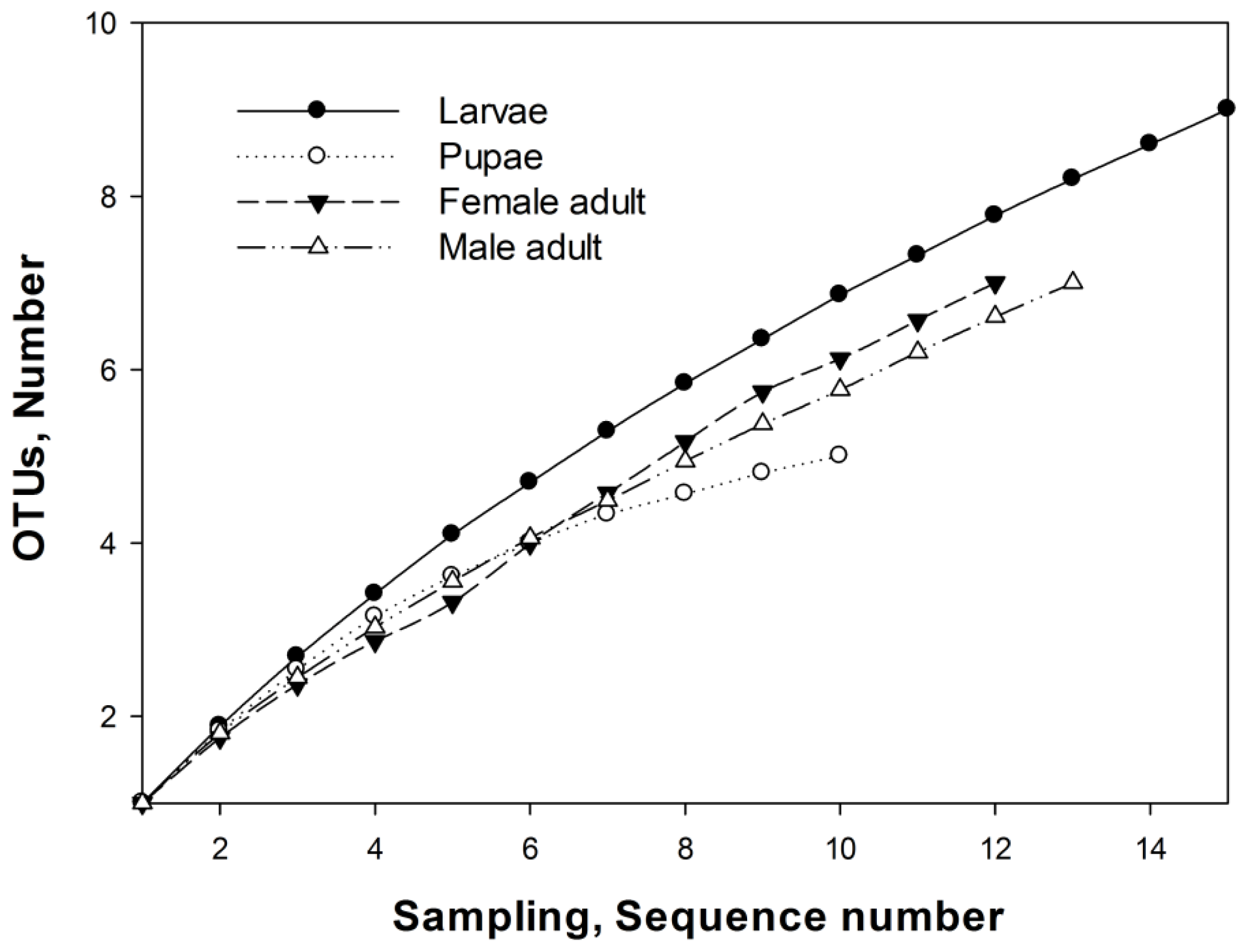
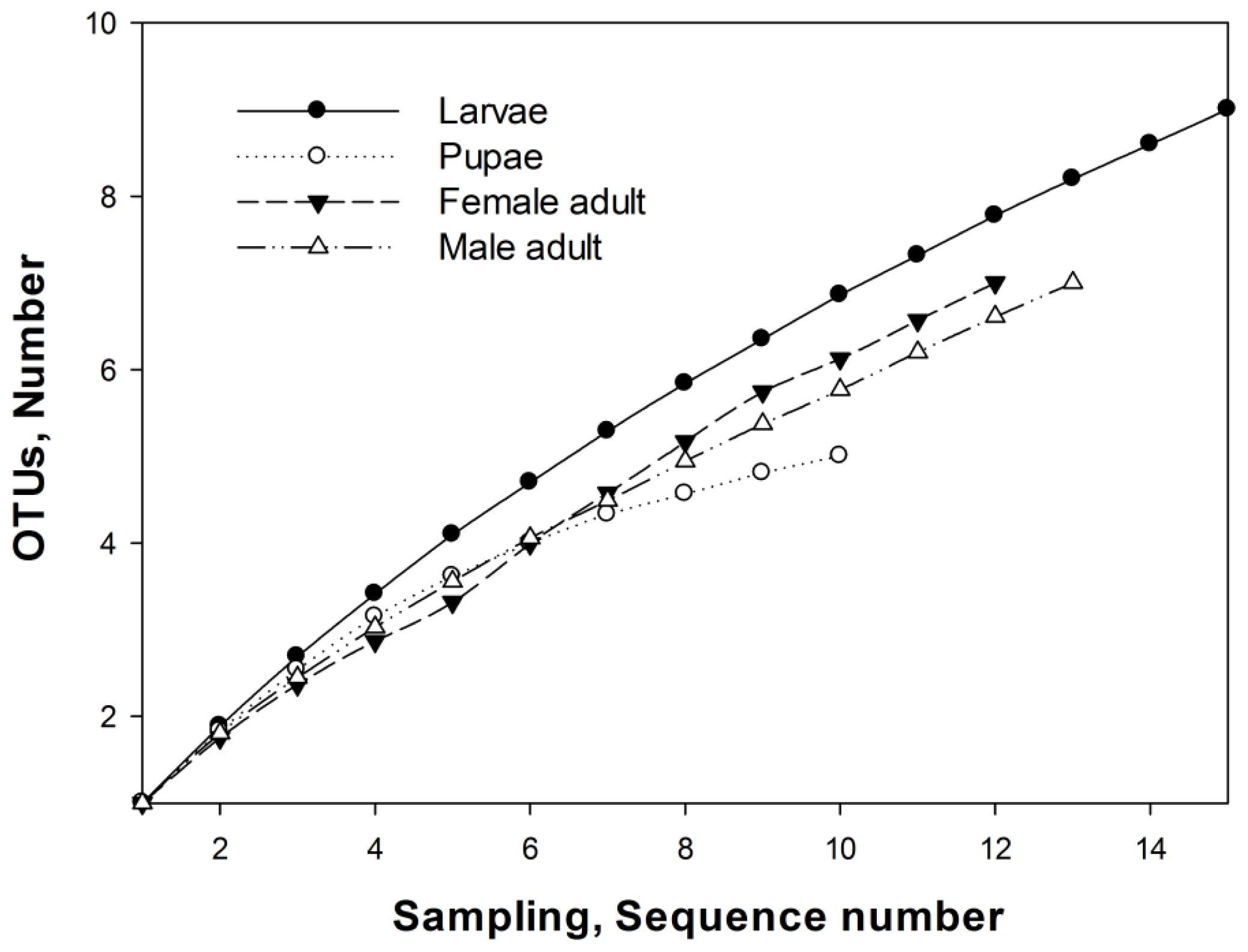
2.3. OTUs
2.4. Bacterial Community Structure
3. Experimental Section
3.1. Insect Collection and Dissection
3.2. Extraction of Bacterial DNA
3.3. Nested PCR
3.4. Denaturing Gradient Gel Electrophoresis (DGGE)
3.5. DGGE Band Identification
3.6. DGGE Band Profile Analysis
3.7. Operational Taxonomic Units and Richness Estimation
4. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lane | D. armandi sample | S | EH | H′ |
|---|---|---|---|---|
| S1 | Larvae | 16 | 0.853886 | 2.367476 |
| S2 | Pupae | 10 | 0.649392 | 1.49528 |
| S3 | Female adult | 12 | 0.854255 | 2.122745 |
| S4 | Male adult | 14 | 0.885312 | 2.336389 |
Acknowledgments
Conflicts of Interest
References
- Haack, R.A.; Slansky, F., Jr. Nutritional Ecology of Wood-Feeding Coleoptera, Lepidoptera, and Hymenoptera. In Nutritional Ecology of Insects, Mites, Spiders, and Related Invertebrates; Slansky, F., Jr., Rodriguez, J., Eds.; Wiley: New York, NY, USA, 1987; pp. 449–486. [Google Scholar]
- Coulson, R.N.; Stark, R.W. Integrated Pest Management of Bark Beetles. In Bark Beetles in North American Conifers: A System for the Study of Evolutionary Biology; Mitton, J.B., Sturgeon, K.B., Eds.; University of Texas Press: Austin, TX, USA, 1982; pp. 315–349. [Google Scholar]
- Cruden, D.L.; Markovetz, A.J. Microbial ecology of the cockroach gut. Annu. Rev. Microbiol 1987, 41, 617–643. [Google Scholar]
- Handelsman, J.; Robinson, C.J.; Raffa, K.F. Microbial Communities in Lepidopteran Guts: From Models to Metagenomics. In The Influence of Cooperative Bacteria on Animal Host Biology; McFall-Ngai, M.J., Henderson, B., Ruby, E.G., Eds.; Cambridge University Press: Cambridge, UK, 2005; pp. 143–145. [Google Scholar]
- Dillon, R.J.; Vennard, C.T.; Charnley, A.K. Pheromones: Exploitation of gut bacteria in the locust. Nature 2000, 403, 851–851. [Google Scholar]
- Moran, N.A.; Tran, P.; Gerardo, N.M. Symbiosis and insect diversification: An ancient symbiont of sap-feeding insects from the bacterial phylum Bacteroidetes. Appl. Environ. Microbiol 2005, 71, 8802–8810. [Google Scholar]
- Bridges, J.R. Nitrogen-fixing bacteria associated with bark beetles. Microb. Ecol 1981, 7, 131–137. [Google Scholar]
- Delalibera, I.; Handelsman, J., Jr.; Raffa, K.F. Contrasts in cellulolytic activities of gut microorganisms between the wood borer, Saperda vestita (Coleoptera: Cerambycidae), and the bark beetles, Ips pini and Dendroctonus frontalis (Coleoptera: Curculionidae). Environ. Entomol 2005, 34, 541–547. [Google Scholar]
- Chen, H.; Tang, M. Spatial and temporal dynamics of bark beetles in Chinese white pine in Qinling Mountains of Shaanxi Province, China. Environ. Entomol 2007, 36, 1124–1130. [Google Scholar]
- Yin, H.F.; Li, Z.L. Economic insect fauna of China. Coleopt. Scolyt 1984, 29, 26–35. [Google Scholar]
- Tang, M.; Chen, H. Effect of symbiotic fungi of Dendroctonus armandi on host trees. Sci. Silvae Sin 1999, 35, 63–66. [Google Scholar]
- Douglas, A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol 2009, 23, 38–47. [Google Scholar]
- Adams, A.S.; Adams, S.M.; Currie, C.R.; Gillette, N.E.; Raffa, K.F. Geographic variation in bacterial communities associated with the Red Turpentine Beetle (Coleoptera: Curculionidae). Environ. Entomol 2010, 39, 406–414. [Google Scholar]
- Nogge, G. Significance of symbionts for the maintenance of an optimal nutritional state for successful reproduction in hematophagous arthropods. Parasitology 1981, 82, 101–104. [Google Scholar]
- Douglas, A.E. Nutritional interactions in insect-microbial symbioses: Aphids and their symbiotic bacteria. Buchnera. Annu. Rev. Entomol 1998, 43, 17–37. [Google Scholar]
- Benemann, J.R. Nitrogen fixation in termites. Science 1973, 181, 164–165. [Google Scholar]
- Dillon, R.J.; Dillon, V.M. The gut bacteria of insects: Nonpathogenic interactions. Annu. Rev. Entomol 2004, 49, 71–92. [Google Scholar]
- Cardoza, Y.J.; Klepzig, K.D.; Raffa, K.F. Bacteria in oral secretions of an endophytic insect inhibit antagonistic fungi. Ecol. Entomol 2006, 31, 636–645. [Google Scholar]
- Geib, S.M.; Filley, T.R.; Hatcher, P.G.; Hoover, K.; Carlson, J.E.; Del Mar Jimenez-Gasco, M.; Nakagawa-Izumi, A.; Sleighter, R.L.; Tien, M. Lignin degradation in wood-feeding insects. Proc. Natl. Acad. Sci. USA 2008, 105, 12932–12937. [Google Scholar]
- Morales-Jiménez, J.; Zúñiga, G.; Villa-Tanaca, L.; Hernández-Rodríguez, C. Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microb. Ecol 2009, 58, 879–891. [Google Scholar]
- Fukatsu, T.; Hosokawa, T. Capsule-transmitted gut symbiotic bacterium of the Japanese common plataspid stinkbug Megacopta punctatissima. Appl. Environ. Microbiol 2002, 68, 389–396. [Google Scholar]
- Funk, D.J.; Bernays, E.A. Geographic variation in host specificity reveals host range evolution in Uroleucon ambrosiae aphids. Ecology 2001, 82, 726–739. [Google Scholar]
- Tsuchida, T.; Koga, R.; Shibao, H.; Matsumoto, T.; Fukatsu, T. Diversity and geographic distribution of secondary endosymbiotic bacteria in natural populations of the pea aphid Acyrthosiphon pisum. Mol. Ecol 2002, 11, 2123–2135. [Google Scholar]
- Hosokawa, T.; Kikuchi, Y.; Meng, X.Y.; Fukatsu, T. The making of symbiont capsule in the plataspid stinkbug Megacopta punctatissima. FEMS Microbiol. Ecol 2005, 54, 471–477. [Google Scholar]
- Broderick, N.A.; Raffa, K.F.; Goodman, R.M.; Handelsman, J. Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl. Environ. Microbiol 2004, 70, 293–300. [Google Scholar]
- Nocker, A.; Burr, M.; Camper, A.K. Genotypic microbial community profiling: A critical technical review. Microb. Ecol 2007, 54, 276–289. [Google Scholar]
- Cocolin, L.; Aggio, D.; Manzano, M.; Cantoni, C.; Comi, G. An application of PCR-DGGE analysis to profile the yeast populations in raw milk. Int. Dairy J 2002, 12, 407–411. [Google Scholar]
- Kuechler, S.M.; Dettner, K.; Kehl, S. Characterization of an obligate intracellular bacterium in the midgut epithelium of the bulrush bug Chilacis typhae (Heteroptera, Lygaeidae, Artheneinae). Appl. Environ. Microbiol 2011, 77, 2869–2876. [Google Scholar]
- Dillon, R.J.; Webster, G.; Weightman, A.J.; Charnley, A.K. Diversity of gut microbiota increases with aging and starvation in the desert locust. Anton. Leeuw. Int. J. G 2010, 97, 69–77. [Google Scholar]
- Lindh, J.M.; Terenius, O.; Faye, I. 16S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl. Environ. Microbiol 2005, 71, 7217–7223. [Google Scholar]
- Lindh, J.M.; Lehane, M.J. The tsetse fly Glossina fuscipes fuscipes (Diptera: Glossina) harbours a surprising diversity of bacteria other than symbionts. Anton. Leeuw. Int. J. G 2011, 99, 711–720. [Google Scholar]
- Weiss, B.; Aksoy, S. Microbiome influences on insect host vector competence. Trends Parasitol 2011, 27, 514–522. [Google Scholar]
- Hongoh, Y. Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosci. Biotechnol. Biochem 2010, 74, 1145–1151. [Google Scholar]
- Breznak, J.A. Intestinal microbiota of termites and other xylophagous insects. Annu. Rev. Microbiol 1982, 36, 323–323. [Google Scholar]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar]
- Delalibera, I., Jr.; Vasanthakumar, A.; Burwitz, B.J.; Schloss, P.D.; Klepzig, K.D.; Handelsman, J.; Raffa, K.F. Composition of the bacterial community in the gut of the pine engraver, Ips pini (Say) (Coleoptera) colonizing red pine. Symbiosis 2007, 43, 97–104. [Google Scholar]
- Winder, R.S.; Macey, D.E.; Cortese, J. Dominant bacteria associated with broods of mountain pine beetle, Dendroctonus ponderosae (Coleoptera: Curculionidae, Scolytinae). J. Entomol. Soc. Brit. Columbia 2010, 107, 43–56. [Google Scholar]
- Morales-Jiménez, J.; de León, A.V.P.; García-Domínguez, A.; Martínez-Romero, E.; Zúñiga, G.; Hernández-Rodríguez, C. Nitrogen-fixing and uricolytic bacteria associated with the gut of Dendroctonus rhizophagus and Dendroctonus valens (Curculionidae: Scolytinae). Microb. Ecol 2013, 66, 200–210. [Google Scholar]
- French, J.R.J.; Turner, G.L.; Bradbury, J.F. Nitrogen fixation by bacteria from the hindgut of termites. Microbiology 1976, 95, 202–206. [Google Scholar]
- Behar, A.; Yuval, B.; Jurkevitch, E. Enterobacteria-mediated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol. Ecol 2005, 14, 2637–2643. [Google Scholar]
- Lilburn, T.G.; Kim, K.S.; Ostrom, N.E.; Byzek, K.R.; Leadbetter, J.R.; Breznak, J.A. Nitrogen fixation by symbiotic and free-living spirochetes. Science 2001, 292, 2495–2498. [Google Scholar]
- Leroy, P.D.; Sabri, A.; Verheggen, F.J.; Francis, F.; Thonart, P.; Haubruge, E. The semiochemically mediated interactions between bacteria and insects. Chemoecology 2011, 21, 113–122. [Google Scholar]
- Peterkova-Koci, K.; Robles-Murguia, M.; Ramalho-Ortigao, M.; Zurek, L. Significance of bacteria in oviposition and larval development of the sand fly Lutzomyia longipalpis. Parasit Vectors 2012, 5, 145. [Google Scholar]
- Morales-Jiménez, J.; Zúñiga, G.; Ramírez-Saad, H.C.; Hernández-Rodríguez, C. Gut-associated bacteria throughout the life cycle of the bark beetle Dendroctonus rhizophagus Thomas and Bright (Curculionidae: Scolytinae) and their cellulolytic activities. Microb. Ecol 2012, 64, 268–278. [Google Scholar]
- Yamoah, E.; Jones, E.E.; Weld, R.J.; Suckling, D.M.; Waipara, N.; Bourdôt, G.W.; Hee, A.K.; Stewart, A. Microbial population and diversity on the exoskeletons of four insect species associated with gorse (Ulex europaeus L.). Aust. J. Entomol 2008, 47, 370–379. [Google Scholar]
- Gai, C.S.; Lacava, P.T.; Quecine, M.C.; Auriac, M.C.; Lopes, J.R.S.; Araújo, W.L.; Miller, T.A.; Azevedo, J.L. Transmission of Methylobacterium mesophilicum by Bucephalogonia xanthophis for paratransgenic control strategy of Citrus Variegated Chlorosis. Microbiology 2009, 47, 448–454. [Google Scholar]
- Patt, T.; Cole, G.; Hanson, R. Methylobacterium, a new genus of facultatively methylotrophic bacteria. Int. J. Syst. Bacteriol 1976, 26, 226–229. [Google Scholar]
- Kuzina, L.W.; Miller, E.D.; Ge, B.; Miller, T.A. Transformation of Enterobacter gergoviae isolated from pink bollworm (Lepidoptera: Gelechiidae) gut with Bacillus thuringiensis toxin. Curr. Microbiol 2002, 44, 1–4. [Google Scholar]
- Lilley, A.K.; Hails, R.S.; Cory, J.S.; Bailey, M.J. The dispersal and establishment of pseudomonas populations in the phyllosphere of sugar beet by phytophagous caterpillars. FEMS Microbiol. Ecol. 1997, 24, 151–157. [Google Scholar]
- Watanabe, K.; Abe, K.; Sato, M. Biological control of an insect pest by gut-colonizing Enterobacter cloacae transformed with ice nucleation gene. J. Appl. Microbiol 2000, 88, 90–97. [Google Scholar]
- Beard, B.C.; Cordon-Rosales, C.; Durvasula, R.V. Bacterial symbionts of the Triatominae and their potential use in control of Chagas disease transmission. Annu. Rev. Entomol 2002, 47, 123–141. [Google Scholar]
- Xu, Z.; Tang, M.; Chen, H.; Ban, Y.; Zhang, H. Microbial community structure in the rhizosphere of Sophora viciifolia grown at a lead and zinc mine of northwest China. Sci. Total Environ 2012, 435, 453–464. [Google Scholar]
- Muyzer, G.; Hottenträger, S.; Teske, A.; Wawer, C. Denaturing Gradient Gel Electrophoresis of PCR-Amplified 16S rDNA—A New Molecular Approach to Analyse the Genetic Diversity of Mixed Microbial Communities. In Molecular Microbial Ecology Manual; Akkermans, A.D.L., Van Elsas, J.D., De Bruijn, F.J., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996; pp. 1–23. [Google Scholar]
- Muyzer, G.; De Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol 1993, 59, 695–700. [Google Scholar]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.; Kulam-Syed-Mohideen, A.; McGarrell, D.; Marsh, T.; Garrity, G. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res 2009, 37, 141–145. [Google Scholar]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 2007, 35, 5–12. [Google Scholar]
- Chun, J.; Lee, J.H.; Jung, Y.; Kim, M.; Kim, S.; Kim, B.K.; Lim, Y.W. EzTaxon: A web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol 2007, 57, 2259–2261. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004, 32, 1792–1797. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol 2011, 28, 2731–2739. [Google Scholar]
- Galand, P.E.; Fritze, H.; Yrjälä, K. Microsite-dependent changes in methanogenic populations in a boreal oligotrophic fen. Environ. Microbiol 2003, 5, 1133–1143. [Google Scholar]
- Lü, D.; Li, Z.; Qin, S.; Ma, H.; Liu, G. Bacterial community structure in the Cerasus sachalinensis Kom. rhizosphere based on the polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) method. Afr. J. Biotechnol 2011, 10, 13430–13438. [Google Scholar]
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microb 2005, 71, 1501–1506. [Google Scholar]
- Chao, A.; Shen, T.J. Nonparametric estimation of Shannon’s index of diversity when there are unseen species in sample. Environ. Ecol. Stat 2003, 10, 429–443. [Google Scholar]
- Gotelli, N.J.; Colwell, R.K. Quantifying biodiversity: Procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett 2001, 4, 379–391. [Google Scholar]
- Colwell, R.K.; Mao, C.X.; Chang, J. Interpolating, extrapolating, and comparing incidence-based species accumulation curves. Ecology 2004, 85, 2717–2727. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hu, X.; Wang, C.; Chen, H.; Ma, J. Differences in the Structure of the Gut Bacteria Communities in Development Stages of the Chinese White Pine Beetle (Dendroctonus armandi). Int. J. Mol. Sci. 2013, 14, 21006-21020. https://doi.org/10.3390/ijms141021006
Hu X, Wang C, Chen H, Ma J. Differences in the Structure of the Gut Bacteria Communities in Development Stages of the Chinese White Pine Beetle (Dendroctonus armandi). International Journal of Molecular Sciences. 2013; 14(10):21006-21020. https://doi.org/10.3390/ijms141021006
Chicago/Turabian StyleHu, Xia, Chunyan Wang, Hui Chen, and Junning Ma. 2013. "Differences in the Structure of the Gut Bacteria Communities in Development Stages of the Chinese White Pine Beetle (Dendroctonus armandi)" International Journal of Molecular Sciences 14, no. 10: 21006-21020. https://doi.org/10.3390/ijms141021006