Impaired Glutathione Synthesis in Neurodegeneration

{kind=link}
{kind=link}
Abstract
:1. Introduction
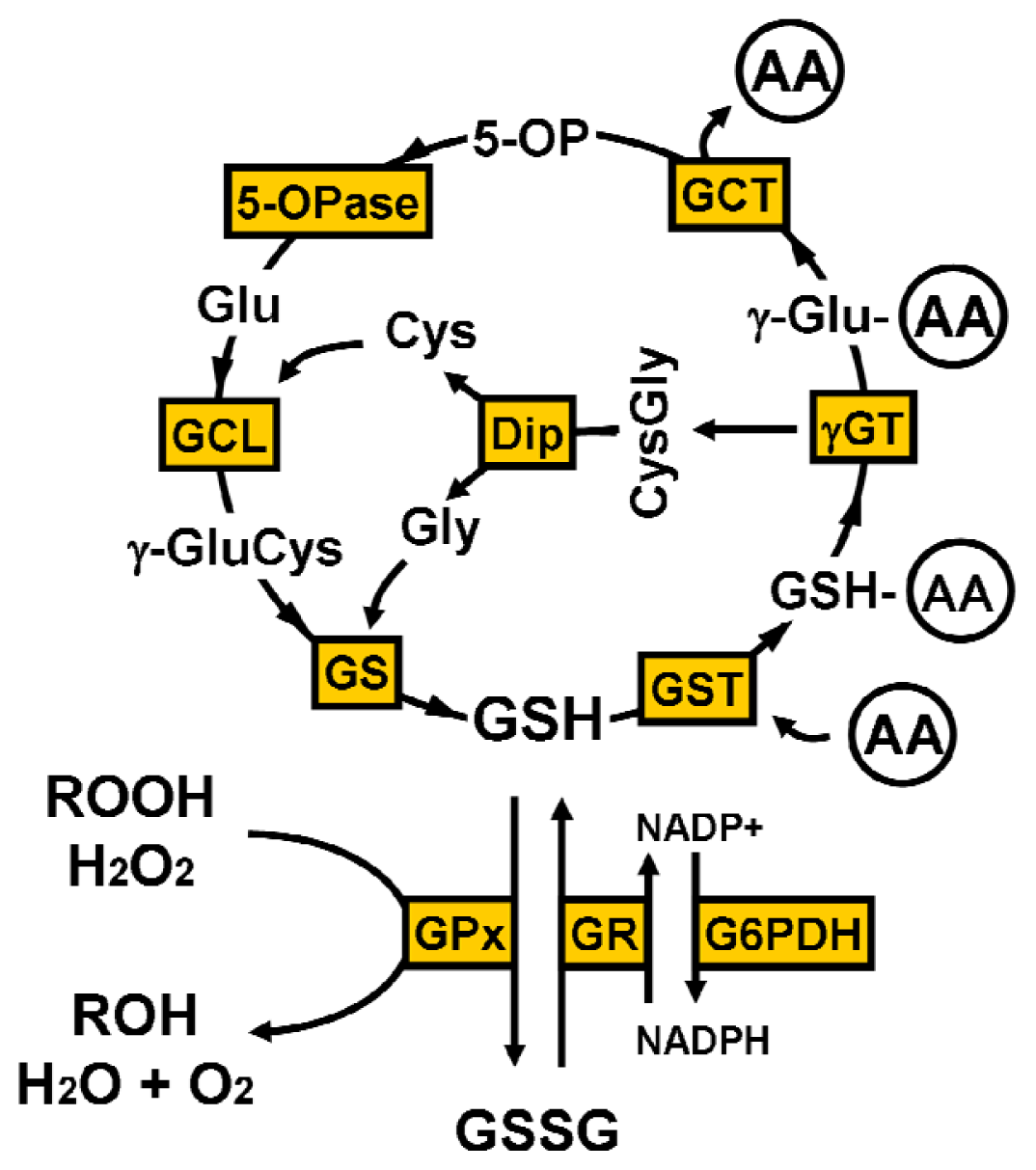
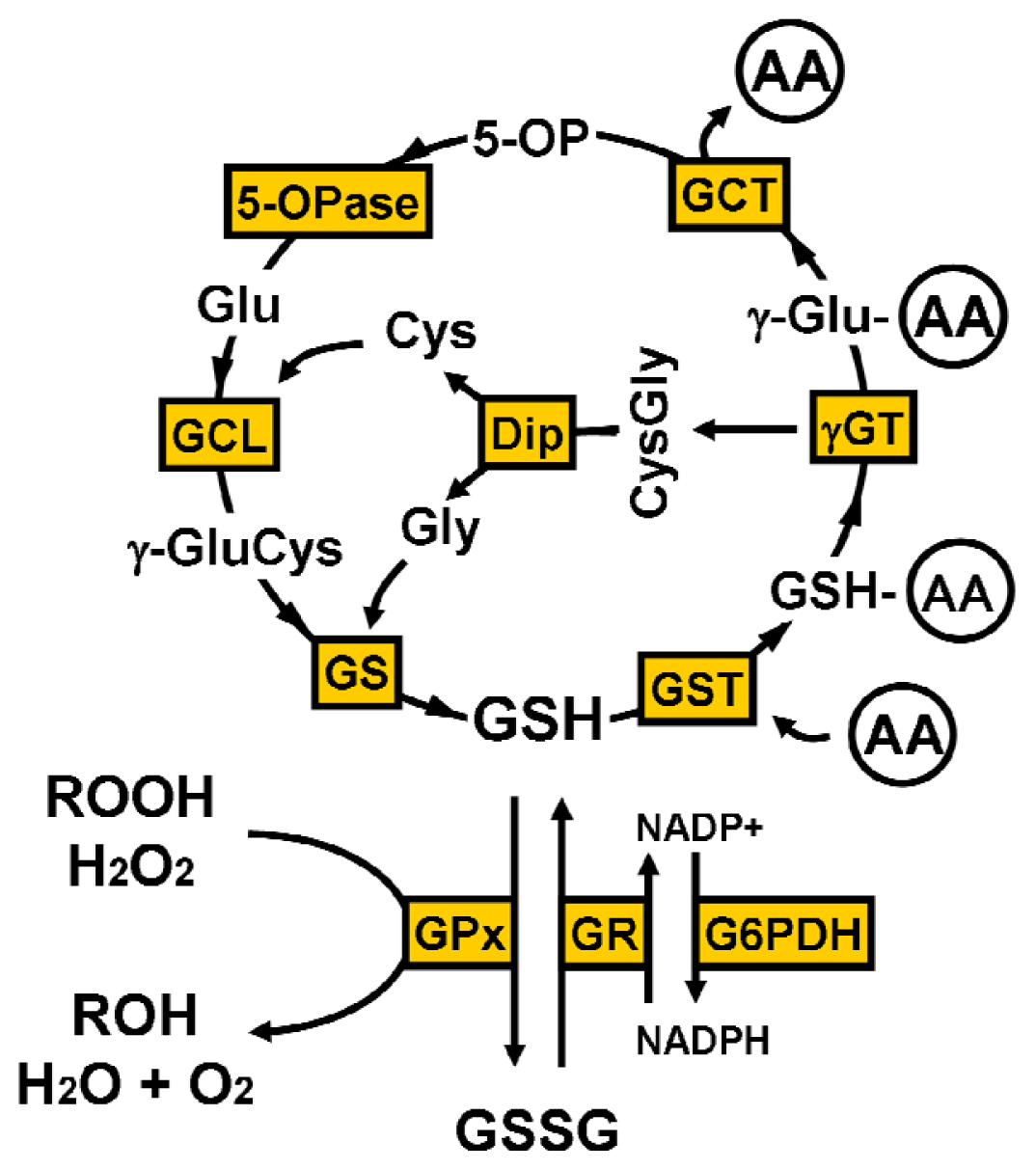
2. Glutathione Synthesis
3. Glutathione in the Brain
3.1. Distribution of GSH in the Brain
3.2. Thiol Source for the Brain
3.3. Thiol Source for Glutathione Synthesis in the Brain
4. Glutathione Function
4.1. γ-Glutamyl Cycle
4.2. Oxidative Stress
4.3. S-Glutathionylation
4.4. Thiol Redox State
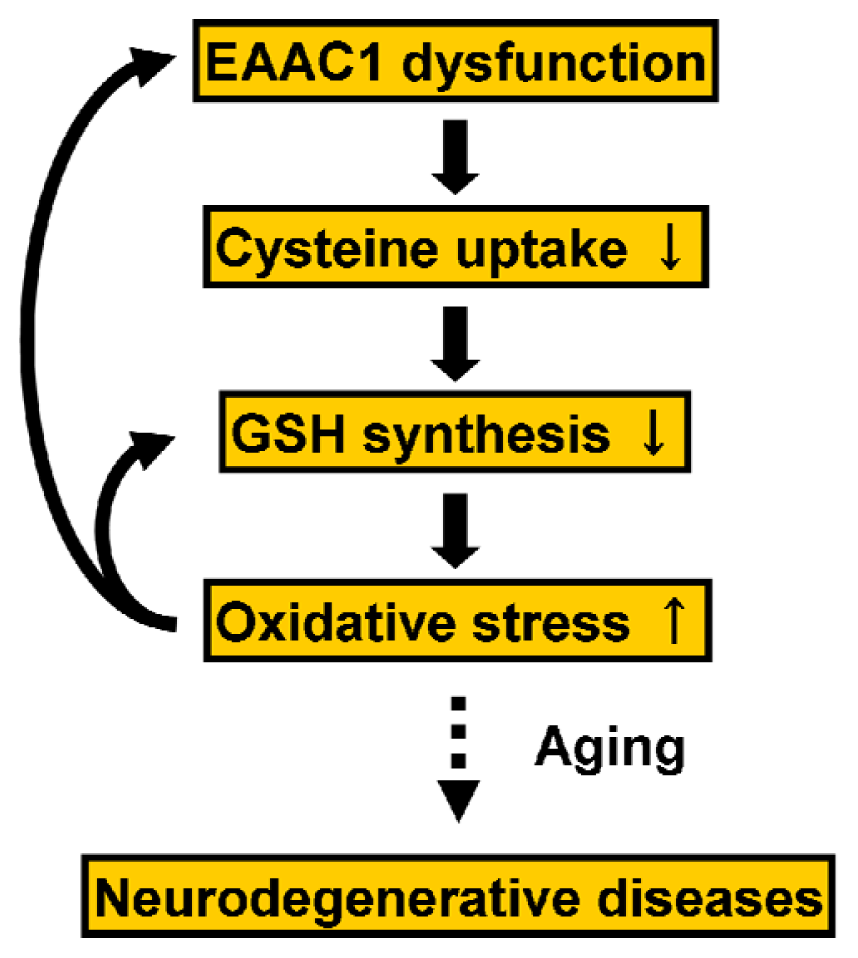
5. Cysteine Uptake for Neuronal Glutathione Synthesis
5.1. The Excitatory Amino Acid Transporters
5.2. Regulation of EAAC1 on Neuronal Glutathione Synthesis
6. Inborn Errors in the GSH-Related Enzymes
6.1. γ-Glutamylcysteine Ligase
6.2. Glutathione Synthetase
6.3. γ-Glutamyl Transpeptidase
6.4. 5-Oxoprolinase
6.5. Membrane-Bound Dipeptidase
7. Disorders of GSH Metabolism in Neurodegenerative Disease
7.1. Alzheimer’s Disease (AD)
7.2. Parkinson’s Disease (PD)
7.3. Amyotrophic Lateral Sclerosis (ALS)
7.4. Progressive Supranuclear Palsy (PSP)
7.5. Huntington’s Disease (HD)
7.6. Multiple Sclerosis (MS)
8. A Potential Approach to Increase GSH Levels in the Brain
9. Conclusions

Conflicts of Interest
References
- De Rey-Pailhade, M.J. Sur un corps d’origine organique hydrogénant le soufre á froid. C. R. Acad. Sci 1888, 106, 1683–1684. [Google Scholar]
- Meister, A. On the discovery of glutathione. Trends Biochem. Sci 1988, 13, 185–188. [Google Scholar]
- Hopkins, F.G. On an autoxidisable constituent of the cell. Biochem. J 1921, 15, 286–305. [Google Scholar]
- Hopkins, F.G. On glutathione: A reinvestigation. J. Biol. Chem 1929, 84, 269–320. [Google Scholar]
- Kendall, E.C.; Mason, H.L.; McKenzie, B.F. A study of glutathione. J. Biol. Chem 1930, 88, 409–423. [Google Scholar]
- Richman, P.G.; Meister, A. Regulation of γ-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J. Biol. Chem 1975, 250, 1422–1426. [Google Scholar]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem 2005, 280, 33766–33774. [Google Scholar]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med 1999, 27, 922–935. [Google Scholar]
- Erecinska, M.; Silver, I.A. Metabolism and role of glutamate in mammalian brain. Prog. Neurobiol 1990, 35, 245–296. [Google Scholar]
- Roux, M.J.; Supplisson, S. Neuronal and glial glycine transporters have different stoichiometries. Neuron 2000, 25, 373–383. [Google Scholar]
- Aoyama, K.; Wang, F.; Matsumura, N.; Kiyonari, H.; Shioi, G.; Tanaka, K.; Kinoshita, C.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Increased neuronal glutathione and neuroprotection in GTRAP3–18-deficient mice. Neurobiol. Dis 2012, 45, 973–982. [Google Scholar]
- Puka-Sundvall, M.; Eriksson, P.; Nilsson, M.; Sandberg, M.; Lehmann, A. Neurotoxicity of cysteine: Interaction with glutamate. Brain Res 1995, 705, 65–70. [Google Scholar]
- Janaky, R.; Varga, V.; Hermann, A.; Saransaari, P.; Oja, S.S. Mechanisms of l-cysteine neurotoxicity. Neurochem. Res 2000, 25, 1397–1405. [Google Scholar]
- Sedlak, J.; Lindsay, R.H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem 1968, 25, 192–205. [Google Scholar]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr 2004, 134, 489–492. [Google Scholar]
- Commandeur, J.N.; Stijntjes, G.J.; Vermeulen, N.P. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol. Rev 1995, 47, 271–330. [Google Scholar]
- Cooper, A.J.; Kristal, B.S. Multiple roles of glutathione in the central nervous system. Biol. Chem 1997, 378, 793–802. [Google Scholar]
- Kang, Y.; Viswanath, V.; Jha, N.; Qiao, X.; Mo, J.Q.; Andersen, J.K. Brain γ-glutamyl cysteine synthetase (GCS) mRNA expression patterns correlate with regional-specific enzyme activities and glutathione levels. J. Neurosci. Res 1999, 58, 436–441. [Google Scholar]
- Anderson, M.E.; Underwood, M.; Bridges, R.J.; Meister, A. Glutathione metabolism at the blood-cerebrospinal fluid barrier. FASEB J 1989, 3, 2527–2531. [Google Scholar]
- Wang, X.F.; Cynader, M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem 2000, 74, 1434–1442. [Google Scholar]
- Makar, T.K.; Nedergaard, M.; Preuss, A.; Gelbard, A.S.; Perumal, A.S.; Cooper, A.J. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: Evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem 1994, 62, 45–53. [Google Scholar]
- Hirrlinger, J.; Gutterer, J.M.; Kussmaul, L.; Hamprecht, B.; Dringen, R. Microglial cells in culture express a prominent glutathione system for the defense against reactive oxygen species. Dev. Neurosci 2000, 22, 384–392. [Google Scholar]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res 2005, 79, 157–165. [Google Scholar]
- Tateishi, N.; Higashi, T.; Naruse, A.; Nakashima, K.; Shiozaki, H. Rat liver glutathione: Possible role as a reservoir of cysteine. J. Nutr 1977, 107, 51–60. [Google Scholar]
- Lauterburg, B.H.; Adams, J.D.; Mitchell, J.R. Hepatic glutathione homeostasis in the rat: Efflux accounts for glutathione turnover. Hepatology 1984, 4, 586–590. [Google Scholar]
- Cornford, E.M.; Braun, L.D.; Crane, P.D.; Oldendorf, W.H. Blood-brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology 1978, 103, 1297–1303. [Google Scholar]
- Lash, L.H.; Jones, D.P. Distribution of oxidized and reduced forms of glutathione and cysteine in rat plasma. Arch. Biochem. Biophys 1985, 240, 583–592. [Google Scholar]
- Ammon, H.P.; Melien, M.C.; Verspohl, E.J. Pharmacokinetics of intravenously administered glutathione in the rat. J. Pharm. Pharmacol 1986, 38, 721–725. [Google Scholar]
- Olney, J.W.; Zorumski, C.; Price, M.T.; Labruyere, J. l-cysteine, a bicarbonate-sensitive endogenous excitotoxin. Science 1990, 248, 596–599. [Google Scholar]
- Wade, L.A.; Brady, H.M. Cysteine and cystine transport at the blood-brain barrier. J. Neurochem 1981, 37, 730–734. [Google Scholar]
- Killian, D.M.; Chikhale, P.J. Predominant functional activity of the large, neutral amino acid transporter (LAT1) isoform at the cerebrovasculature. Neurosci. Lett 2001, 306, 1–4. [Google Scholar]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem 1999, 274, 11455–11458. [Google Scholar]
- Pow, D.V. Visualising the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia 2001, 34, 27–38. [Google Scholar]
- Qin, S.; Colin, C.; Hinners, I.; Gervais, A.; Cheret, C.; Mallat, M. System xc − and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-β peptide 1–40. J. Neurosci 2006, 26, 3345–3356. [Google Scholar]
- Dringen, R.; Kranich, O.; Loschmann, P.A.; Hamprecht, B. Use of dipeptides for the synthesis of glutathione by astroglia-rich primary cultures. J. Neurochem 1997, 69, 868–874. [Google Scholar]
- Dringen, R.; Hamprecht, B.; Broer, S. The peptide transporter PepT2 mediates the uptake of the glutathione precursor CysGly in astroglia-rich primary cultures. J. Neurochem 1998, 71, 388–393. [Google Scholar]
- Dringen, R.; Hamprecht, B. Glutathione restoration as indicator for cellular metabolism of astroglial cells. Dev. Neurosci 1998, 20, 401–407. [Google Scholar]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem 2003, 384, 505–516. [Google Scholar]
- Dringen, R.; Gutterer, J.M.; Gros, C.; Hirrlinger, J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J. Neurosci. Res 2001, 66, 1003–1008. [Google Scholar]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci 1999, 19, 562–569. [Google Scholar]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev 1979, 59, 527–605. [Google Scholar]
- Staniek, K.; Nohl, H. Are mitochondria a permanent source of reactive oxygen species? Biochim. Biophys. Acta 2000, 1460, 268–275. [Google Scholar]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem 2002, 277, 44784–44790. [Google Scholar]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cocheme, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci 2012, 125, 801–806. [Google Scholar]
- Girotti, A.W. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res 1998, 39, 1529–1542. [Google Scholar]
- Trepanier, G.; Furling, D.; Puymirat, J.; Mirault, M.E. Immunocytochemical localization of seleno-glutathione peroxidase in the adult mouse brain. Neuroscience 1996, 75, 231–243. [Google Scholar]
- Arthur, J.R. The glutathione peroxidases. Cell. Mol. Life Sci 2000, 57, 1825–1835. [Google Scholar]
- Power, J.H.; Blumbergs, P.C. Cellular glutathione peroxidase in human brain: Cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol 2009, 117, 63–73. [Google Scholar]
- Yang, Y.; Sharma, R.; Sharma, A.; Awasthi, S.; Awasthi, Y.C. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim. Pol 2003, 50, 319–336. [Google Scholar]
- Winterbourn, C.C.; Metodiewa, D. The reaction of superoxide with reduced glutathione. Arch. Biochem. Biophys 1994, 314, 284–290. [Google Scholar]
- Hogg, N.; Singh, R.J.; Kalyanaraman, B. The role of glutathione in the transport and catabolism of nitric oxide. FEBS Lett 1996, 382, 223–228. [Google Scholar]
- Dringen, R.; Gutterer, J.M. Glutathione reductase from bovine brain. Methods Enzymol 2002, 348, 281–288. [Google Scholar]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem 1999, 72, 2523–2530. [Google Scholar]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol 2005, 45, 51–88. [Google Scholar]
- Leier, I.; Jedlitschky, G.; Buchholz, U.; Center, M.; Cole, S.P.; Deeley, R.G.; Keppler, D. ATP-dependent glutathione disulphide transport mediated by the MRP gene-encoded conjugate export pump. Biochem. J 1996, 314, 433–437. [Google Scholar]
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Aspects Med 2009, 30, 13–28. [Google Scholar]
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem 1997, 272, 16644–16651. [Google Scholar]
- Malinski, T.; Bailey, F.; Zhang, Z.G.; Chopp, M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab 1993, 13, 355–358. [Google Scholar]
- Cherian, L.; Goodman, J.C.; Robertson, C.S. Brain nitric oxide changes after controlled cortical impact injury in rats. J. Neurophysiol 2000, 83, 2171–2178. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 2007, 87, 315–424. [Google Scholar]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov 2007, 6, 662–680. [Google Scholar]
- Beckman, J.S. Peroxynitrite versus hydroxyl radical: The role of nitric oxide in superoxide-dependent cerebral injury. Ann. N. Y. Acad. Sci 1994, 738, 69–75. [Google Scholar]
- Koppenol, W.H. The basic chemistry of nitrogen monoxide and peroxynitrite. Free Radic. Biol. Med 1998, 25, 385–391. [Google Scholar]
- Yamakura, F.; Taka, H.; Fujimura, T.; Murayama, K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J. Biol. Chem 1998, 273, 14085–14089. [Google Scholar]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci 2008, 108, 227–238. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J 1984, 219, 1–14. [Google Scholar]
- Sies, H. Strategies of antioxidant defense. Eur. J. Biochem 1993, 215, 213–219. [Google Scholar]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem 2000, 267, 4928–4944. [Google Scholar]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med 2006, 10, 389–406. [Google Scholar]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-Glutathionylation: From redox regulation of protein functions to human diseases. J. Cell. Mol. Med 2004, 8, 201–212. [Google Scholar]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem 2000, 267, 6102–6109. [Google Scholar]
- Berndt, C.; Lillig, C.H.; Holmgren, A. Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim. Biophys. Acta 2008, 1783, 641–650. [Google Scholar]
- Gilbert, H.F. Redox control of enzyme activities by thiol/disulfide exchange. Methods Enzymol 1984, 107, 330–351. [Google Scholar]
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol 1995, 251, 8–28. [Google Scholar]
- Arrigo, A.P. Gene expression and the thiol redox state. Free Radic. Biol. Med 1999, 27, 936–944. [Google Scholar]
- Klatt, P.; Molina, E.P.; de Lacoba, M.G.; Padilla, C.A.; Martinez-Galesteo, E.; Barcena, J.A.; Lamas, S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J 1999, 13, 1481–1490. [Google Scholar]
- Pineda-Molina, E.; Klatt, P.; Vazquez, J.; Marina, A.; Garcia de Lacoba, M.; Perez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-κB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar]
- Fratelli, M.; Goodwin, L.O.; Orom, U.A.; Lombardi, S.; Tonelli, R.; Mengozzi, M.; Ghezzi, P. Gene expression profiling reveals a signaling role of glutathione in redox regulation. Proc. Natl. Acad. Sci. USA 2005, 102, 13998–14003. [Google Scholar]
- Poot, M.; Teubert, H.; Rabinovitch, P.S.; Kavanagh, T.J. De novo synthesis of glutathione is required for both entry into and progression through the cell cycle. J. Cell. Physiol 1995, 163, 555–560. [Google Scholar]
- Markovic, J.; Borras, C.; Ortega, A.; Sastre, J.; Vina, J.; Pallardo, F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. J. Biol. Chem 2007, 282, 20416–20424. [Google Scholar]
- Voehringer, D.W. BCL-2 and glutathione: Alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic. Biol. Med 1999, 27, 945–950. [Google Scholar]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 MAP kinase pathway. FASEB J 2003, 17, 64–66. [Google Scholar] [Green Version]
- Sagara, J.I.; Miura, K.; Bannai, S. Maintenance of neuronal glutathione by glial cells. J. Neurochem 1993, 61, 1672–1676. [Google Scholar]
- Kranich, O.; Hamprecht, B.; Dringen, R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci. Lett 1996, 219, 211–214. [Google Scholar]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol 2001, 65, 1–105. [Google Scholar]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res 2001, 902, 156–163. [Google Scholar]
- Kanai, Y.; Hediger, M.A. The glutamate and neutral amino acid transporter family: Physiological and pharmacological implications. Eur. J. Pharmacol 2003, 479, 237–247. [Google Scholar]
- Had-Aissouni, L. Maintenance of antioxidant defenses of brain cells: Plasma membrane glutamate transporters and beyond. Amino Acids 2012, 42, 159–161. [Google Scholar]
- Had-Aissouni, L. Toward a new role for plasma membrane sodium-dependent glutamate transporters of astrocytes: Maintenance of antioxidant defenses beyond extracellular glutamate clearance. Amino Acids 2012, 42, 181–197. [Google Scholar]
- Zerangue, N.; Kavanaugh, M.P. Flux coupling in a neuronal glutamate transporter. Nature 1996, 383, 634–637. [Google Scholar]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar]
- Coco, S.; Verderio, C.; Trotti, D.; Rothstein, J.D.; Volterra, A.; Matteoli, M. Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur. J. Neurosci 1997, 9, 1902–1910. [Google Scholar]
- Shashidharan, P.; Huntley, G.W.; Murray, J.M.; Buku, A.; Moran, T.; Walsh, M.J.; Morrison, J.H.; Plaitakis, A. Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res 1997, 773, 139–148. [Google Scholar]
- Holmseth, S.; Dehnes, Y.; Huang, Y.H.; Follin-Arbelet, V.V.; Grutle, N.J.; Mylonakou, M.N.; Plachez, C.; Zhou, Y.; Furness, D.N.; Bergles, D.E.; et al. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J. Neurosci 2012, 32, 6000–6013. [Google Scholar]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar]
- Grewer, C.; Rauen, T. Electrogenic glutamate transporters in the CNS: Molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J. Membr. Biol 2005, 203, 1–20. [Google Scholar]
- Zerangue, N.; Kavanaugh, M.P. Interaction of l-cysteine with a human excitatory amino acid transporter. J. Physiol 1996, 493, 419–423. [Google Scholar]
- Himi, T.; Ikeda, M.; Yasuhara, T.; Nishida, M.; Morita, I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm 2003, 110, 1337–1348. [Google Scholar]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci 2006, 9, 119–126. [Google Scholar]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; Swanson, R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1−/− mouse. Ann. Neurol 2011, 69, 509–520. [Google Scholar]
- Fournier, K.M.; Gonzalez, M.I.; Robinson, M.B. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J. Biol. Chem 2004, 279, 34505–34513. [Google Scholar]
- Ruggiero, A.M.; Liu, Y.; Vidensky, S.; Maier, S.; Jung, E.; Farhan, H.; Robinson, M.B.; Sitte, H.; Rothstein, J.D. The ER exit of glutamate transporter is regulated by the inducible mammalian Yip6b/GTRAP3–18 protein. J. Biol. Chem 2008, 283, 6175–6183. [Google Scholar]
- Watabe, M.; Aoyama, K.; Nakaki, T. Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3–18 (GTRAP3–18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol. Pharmacol 2007, 72, 1103–1110. [Google Scholar]
- Watabe, M.; Aoyama, K.; Nakaki, T. A dominant role of GTRAP3–18 in neuronal glutathione synthesis. J. Neurosci 2008, 28, 9404–9413. [Google Scholar]
- Aoyama, K.; Nakaki, T. Inhibition of GTRAP3–18 may increase neuroprotective glutathione (GSH) synthesis. Int. J. Mol. Sci 2012, 13, 12017–12035. [Google Scholar]
- Aoyama, K.; Nakaki, T. Neuroprotective properties of the excitatory amino acid carrier 1 (EAAC1). Amino Acids 2013, 45, 133–142. [Google Scholar]
- Dalton, T.P.; Dieter, M.Z.; Yang, Y.; Shertzer, H.G.; Nebert, D.W. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: Embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem. Biophys. Res. Commun 2000, 279, 324–329. [Google Scholar]
- Yang, Y.; Dieter, M.Z.; Chen, Y.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem 2002, 277, 49446–49452. [Google Scholar]
- Chen, Y.; Johansson, E.; Fan, Y.; Shertzer, H.G.; Vasiliou, V.; Nebert, D.W.; Dalton, T.P. Early onset senescence occurs when fibroblasts lack the glutamate-cysteine ligase modifier subunit. Free Radic. Biol. Med 2009, 47, 410–418. [Google Scholar]
- Ristoff, E.; Larsson, A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis 2007, 2, 16. [Google Scholar]
- Winkler, A.; Njalsson, R.; Carlsson, K.; Elgadi, A.; Rozell, B.; Abraham, L.; Ercal, N.; Shi, Z.Z.; Lieberman, M.W.; Larsson, A.; et al. Glutathione is essential for early embryogenesis—Analysis of a glutathione synthetase knockout mouse. Biochem. Biophys. Res. Commun 2011, 412, 121–126. [Google Scholar]
- Choi, I.Y.; Gruetter, R. Dynamic or inert metabolism? Turnover of N-acetyl aspartate and glutathione from d-[1-13C]glucose in the rat brain in vivo. J. Neurochem. 2004, 91, 778–787. [Google Scholar]
- Jenner, P. Oxidative damage in neurodegenerative disease. Lancet 1994, 344, 796–798. [Google Scholar]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol 2003, 53, S26–S36. [Google Scholar]
- Maher, P. The effects of stress and aging on glutathione metabolism. Ageing Res. Rev 2005, 4, 288–314. [Google Scholar]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev 1998, 78, 547–581. [Google Scholar]
- Jain, A.; Martensson, J.; Stole, E.; Auld, P.A.; Meister, A. Glutathione deficiency leads to mitochondrial damage in brain. Proc. Natl. Acad. Sci. USA 1991, 88, 1913–1917. [Google Scholar]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem 2000, 267, 4912–4916. [Google Scholar]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem 2000, 267, 4904–4911. [Google Scholar]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev 1997, 25, 335–358. [Google Scholar]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med 2004, 10, S18–S25. [Google Scholar]
- Mandal, P.K.; Tripathi, M.; Sugunan, S. Brain oxidative stress: Detection and mapping of anti-oxidant marker “Glutathione” in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun 2012, 417, 43–48. [Google Scholar]
- Metcalfe, T.; Bowen, D.M.; Muller, D.P. Vitamin E concentrations in human brain of patients with Alzheimer’s disease, fetuses with Down’s syndrome, centenarians, and controls. Neurochem. Res 1989, 14, 1209–1212. [Google Scholar]
- Paraskevas, G.P.; Kapaki, E.; Libitaki, G.; Zournas, C.; Segditsa, I.; Papageorgiou, C. Ascorbate in healthy subjects, amyotrophic lateral sclerosis and Alzheimer’s disease. Acta Neurol. Scand 1997, 96, 88–90. [Google Scholar]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology 1998, 51, 1562–1566. [Google Scholar]
- Casado, A.; Encarnacion Lopez-Fernandez, M.; Concepcion Casado, M.; de la Torre, R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem. Res 2008, 33, 450–458. [Google Scholar]
- Spalletta, G.; Bernardini, S.; Bellincampi, L.; Federici, G.; Trequattrini, A.; Ciappi, F.; Bria, P.; Caltagirone, C.; Bossu, P. Glutathione S-transferase P1 and T1 gene polymorphisms predict longitudinal course and age at onset of Alzheimer disease. Am. J. Geriatr. Psychiatry 2007, 15, 879–887. [Google Scholar]
- Paz-y-Mino, C.; Carrera, C.; Lopez-Cortes, A.; Munoz, M.J.; Cumbal, N.; Castro, B.; Cabrera, A.; Sanchez, M.E. Genetic polymorphisms in apolipoprotein E and glutathione peroxidase 1 genes in the Ecuadorian population affected with Alzheimer’s disease. Am. J. Med. Sci 2010, 340, 373–377. [Google Scholar]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar]
- Ramassamy, C.; Averill, D.; Beffert, U.; Theroux, L.; Lussier-Cacan, S.; Cohn, J.S.; Christen, Y.; Schoofs, A.; Davignon, J.; Poirier, J. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol. Dis 2000, 7, 23–37. [Google Scholar]
- Petersen, R.C. Mild cognitive impairment: Transition between aging and Alzheimer’s disease. Neurologia 2000, 15, 93–101. [Google Scholar]
- Sultana, R.; Piroddi, M.; Galli, F.; Butterfield, D.A. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem. Res 2008, 33, 2540–2546. [Google Scholar]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol 2007, 8, 101–112. [Google Scholar]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis 2013, 36, 197–209. [Google Scholar]
- Duerson, K.; Woltjer, R.L.; Mookherjee, P.; Leverenz, J.B.; Montine, T.J.; Bird, T.D.; Pow, D.V.; Rauen, T.; Cook, D.G. Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer’s disease patients. Brain Pathol 2009, 19, 267–278. [Google Scholar]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol 2010, 69, 155–167. [Google Scholar]
- Frank, C.; Pari, G.; Rossiter, J.P. Approach to diagnosis of Parkinson disease. Can. Fam. Physician 2006, 52, 862–868. [Google Scholar]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet 2007, 16, R183–R194. [Google Scholar]
- Khandhar, S.M.; Marks, W.J. Epidemiology of Parkinson’s disease. Dis. Mon 2007, 53, 200–205. [Google Scholar]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord 2011, 26, 1049–1055. [Google Scholar]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol 1994, 36, 348–355. [Google Scholar]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem 1989, 52, 515–520. [Google Scholar]
- Dexter, D.T.; Ward, R.J.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Peters, T.J.; Jenner, P.; Marsden, C.D. α-Tocopherol levels in brain are not altered in Parkinson’s disease. Ann. Neurol 1992, 32, 591–593. [Google Scholar]
- Dexter, D.T.; Sian, J.; Rose, S.; Hindmarsh, J.G.; Mann, V.M.; Cooper, J.M.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Schapira, A.H.; et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann. Neurol 1994, 35, 38–44. [Google Scholar]
- Paik, S.R.; Lee, D.; Cho, H.J.; Lee, E.N.; Chang, C.S. Oxidized glutathione stimulated the amyloid formation of α-synuclein. FEBS Lett 2003, 537, 63–67. [Google Scholar]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med 2012, 2, a008888. [Google Scholar]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53→Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar]
- Vali, S.; Chinta, S.J.; Peng, J.; Sultana, Z.; Singh, N.; Sharma, P.; Sharada, S.; Andersen, J.K.; Bharath, M.M. Insights into the effects of α-synuclein expression and proteasome inhibition on glutathione metabolism through a dynamic in silico model of Parkinson’s disease: Validation by cell culture data. Free Radic. Biol. Med 2008, 45, 1290–1301. [Google Scholar]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar]
- Bossy-Wetzel, E.; Schwarzenbacher, R.; Lipton, S.A. Molecular pathways to neurodegeneration. Nat. Med 2004, 10, S2–S9. [Google Scholar]
- Wong, E.S.; Tan, J.M.; Wang, C.; Zhang, Z.; Tay, S.P.; Zaiden, N.; Ko, H.S.; Dawson, V.L.; Dawson, T.M.; Lim, K.L. Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J. Biol. Chem 2007, 282, 12310–12318. [Google Scholar]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener 2011, 6, 34. [Google Scholar]
- Casarejos, M.J.; Solano, R.M.; Rodriguez-Navarro, J.A.; Gomez, A.; Perucho, J.; Castano, J.G.; Garcia de Yebenes, J.; Mena, M.A. Parkin deficiency increases the resistance of midbrain neurons and glia to mild proteasome inhibition: The role of autophagy and glutathione homeostasis. J. Neurochem 2009, 110, 1523–1537. [Google Scholar]
- Zhou, W.; Zhu, M.; Wilson, M.A.; Petsko, G.A.; Fink, A.L. The oxidation state of DJ-1 regulates its chaperone activity toward α-synuclein. J. Mol. Biol 2006, 356, 1036–1048. [Google Scholar]
- Zhou, W.; Freed, C.R. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T α-synuclein toxicity. J. Biol. Chem 2005, 280, 43150–43158. [Google Scholar]
- Choi, J.; Sullards, M.C.; Olzmann, J.A.; Rees, H.D.; Weintraub, S.T.; Bostwick, D.E.; Gearing, M.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem 2006, 281, 10816–10824. [Google Scholar]
- Plaitakis, A.; Shashidharan, P. Glutamate transport and metabolism in dopaminergic neurons of substantia nigra: Implications for the pathogenesis of Parkinson’s disease. J. Neurol 2000, 247, II25–II35. [Google Scholar]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci 1998, 19, 328–334. [Google Scholar]
- Nafia, I.; Re, D.B.; Masmejean, F.; Melon, C.; Kachidian, P.; Kerkerian-Le Goff, L.; Nieoullon, A.; Had-Aissouni, L. Preferential vulnerability of mesencephalic dopamine neurons to glutamate transporter dysfunction. J. Neurochem 2008, 105, 484–496. [Google Scholar]
- Aoyama, K.; Matsumura, N.; Watabe, M.; Nakaki, T. Oxidative stress on EAAC1 is involved in MPTP-induced glutathione depletion and motor dysfunction. Eur. J. Neurosci 2008, 27, 20–30. [Google Scholar]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochem. Int 2008, 52, 1284–1289. [Google Scholar]
- Lanius, R.A.; Krieger, C.; Wagey, R.; Shaw, C.A. Increased [35S]glutathione binding sites in spinal cords from patients with sporadic amyotrophic lateral sclerosis. Neurosci. Lett 1993, 163, 89–92. [Google Scholar]
- Usarek, E.; Gajewska, B.; Kazmierczak, B.; Kuzma, M.; Dziewulska, D.; Baranczyk-Kuzma, A. A study of glutathione S-transferase pi expression in central nervous system of subjects with amyotrophic lateral sclerosis using RNA extraction from formalin-fixed, paraffin-embedded material. Neurochem. Res 2005, 30, 1003–1007. [Google Scholar]
- Rothstein, J.D.; van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol 1995, 38, 73–84. [Google Scholar]
- Bristol, L.A.; Rothstein, J.D. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann. Neurol 1996, 39, 676–679. [Google Scholar]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar]
- Azbill, R.D.; Mu, X.; Springer, J.E. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res 2000, 871, 175–180. [Google Scholar]
- Frizzo, M.E.; Dall’Onder, L.P.; Dalcin, K.B.; Souza, D.O. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell. Mol. Neurobiol 2004, 24, 123–128. [Google Scholar]
- Deng, Y.; Xu, Z.F.; Liu, W.; Xu, B.; Yang, H.B.; Wei, Y.G. Riluzole-triggered GSH synthesis via activation of glutamate transporters to antagonize methylmercury-induced oxidative stress in rat cerebral cortex. Oxid. Med. Cell. Longev 2012, 2012, 534705. [Google Scholar]
- Fitzmaurice, P.S.; Ang, L.; Guttman, M.; Rajput, A.H.; Furukawa, Y.; Kish, S.J. Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov. Disord 2003, 18, 969–976. [Google Scholar]
- Friguet, B.; Stadtman, E.R.; Szweda, L.I. Modification of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Formation of cross-linked protein that inhibits the multicatalytic protease. J. Biol. Chem 1994, 269, 21639–21643. [Google Scholar]
- Kinter, M.; Roberts, R.J. Glutathione consumption and glutathione peroxidase inactivation in fibroblast cell lines by 4-hydroxy-2-nonenal. Free Radic. Biol. Med 1996, 21, 457–462. [Google Scholar]
- Aoyama, K.; Matsubara, K.; Kobayashi, S. Aging and oxidative stress in progressive supranuclear palsy. Eur. J. Neurol 2006, 13, 89–92. [Google Scholar]
- Albers, D.S.; Augood, S.J. New insights into progressive supranuclear palsy. Trends Neurosci 2001, 24, 347–353. [Google Scholar]
- Klepac, N.; Relja, M.; Klepac, R.; Hecimovic, S.; Babic, T.; Trkulja, V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol 2007, 254, 1676–1683. [Google Scholar]
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; Difiglia, M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci 2010, 30, 4552–4561. [Google Scholar]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci 2008, 31, 247–269. [Google Scholar]
- Dutta, R.; Trapp, B.D. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog. Neurobiol 2011, 93, 1–12. [Google Scholar]
- Srinivasan, R.; Ratiney, H.; Hammond-Rosenbluth, K.E.; Pelletier, D.; Nelson, S.J. MR spectroscopic imaging of glutathione in the white and gray matter at 7 T with an application to multiple sclerosis. Magn. Reson. Imaging 2010, 28, 163–170. [Google Scholar]
- Choi, I.Y.; Lee, S.P.; Denney, D.R.; Lynch, S.G. Lower levels of glutathione in the brains of secondary progressive multiple sclerosis patients measured by 1H magnetic resonance chemical shift imaging at 3 T. Mult. Scler 2011, 17, 289–296. [Google Scholar]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. The role of oxidative stress in the pathogenesis of multiple sclerosis: The need for effective antioxidant therapy. J. Neurol 2004, 251, 261–268. [Google Scholar]
- Wendel, A.; Cikryt, P. The level and half-life of glutathione in human plasma. FEBS Lett 1980, 120, 209–211. [Google Scholar]
- Wendel, A.; Jaeschke, H. Drug-induced lipid peroxidation in mice-III. Glutathione content of liver, kidney and spleen after intravenous administration of free and liposomally entrapped glutathione. Biochem. Pharmacol 1982, 31, 3607–3611. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aoyama, K.; Nakaki, T. Impaired Glutathione Synthesis in Neurodegeneration. Int. J. Mol. Sci. 2013, 14, 21021-21044. https://doi.org/10.3390/ijms141021021
Aoyama K, Nakaki T. Impaired Glutathione Synthesis in Neurodegeneration. International Journal of Molecular Sciences. 2013; 14(10):21021-21044. https://doi.org/10.3390/ijms141021021
Chicago/Turabian StyleAoyama, Koji, and Toshio Nakaki. 2013. "Impaired Glutathione Synthesis in Neurodegeneration" International Journal of Molecular Sciences 14, no. 10: 21021-21044. https://doi.org/10.3390/ijms141021021