Regulation of Ovarian Cancer Stem Cells or Tumor-Initiating Cells


Abstract
:1. Introduction
2. Ovarian CSC/TICs
2.1. Concept of CSC/TICs
2.2. Ovarian CSC/TICs
2.2.1. CD44, CD117, and CD24
2.2.2. CD133 and ALDH1
3. Genes and Pathways Regulating Ovarian CSC/TICs
3.1. Pathways Involved in the Regulation of Ovarian CSC/TICs
3.2. Genes Involved in the Regulation of Ovarian CSC/TICs
4. MiRNAs Regulating Ovarian CSC/TICs
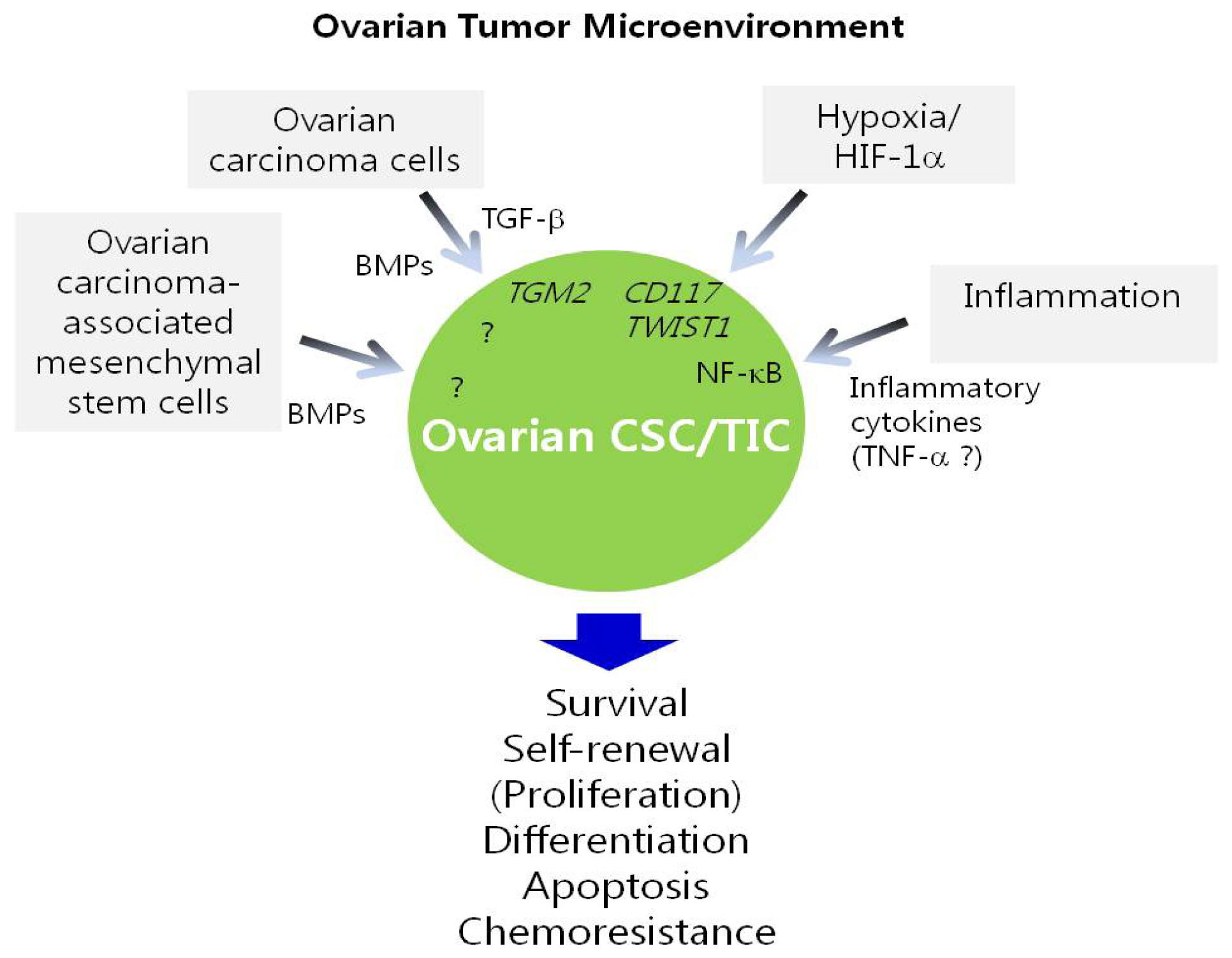
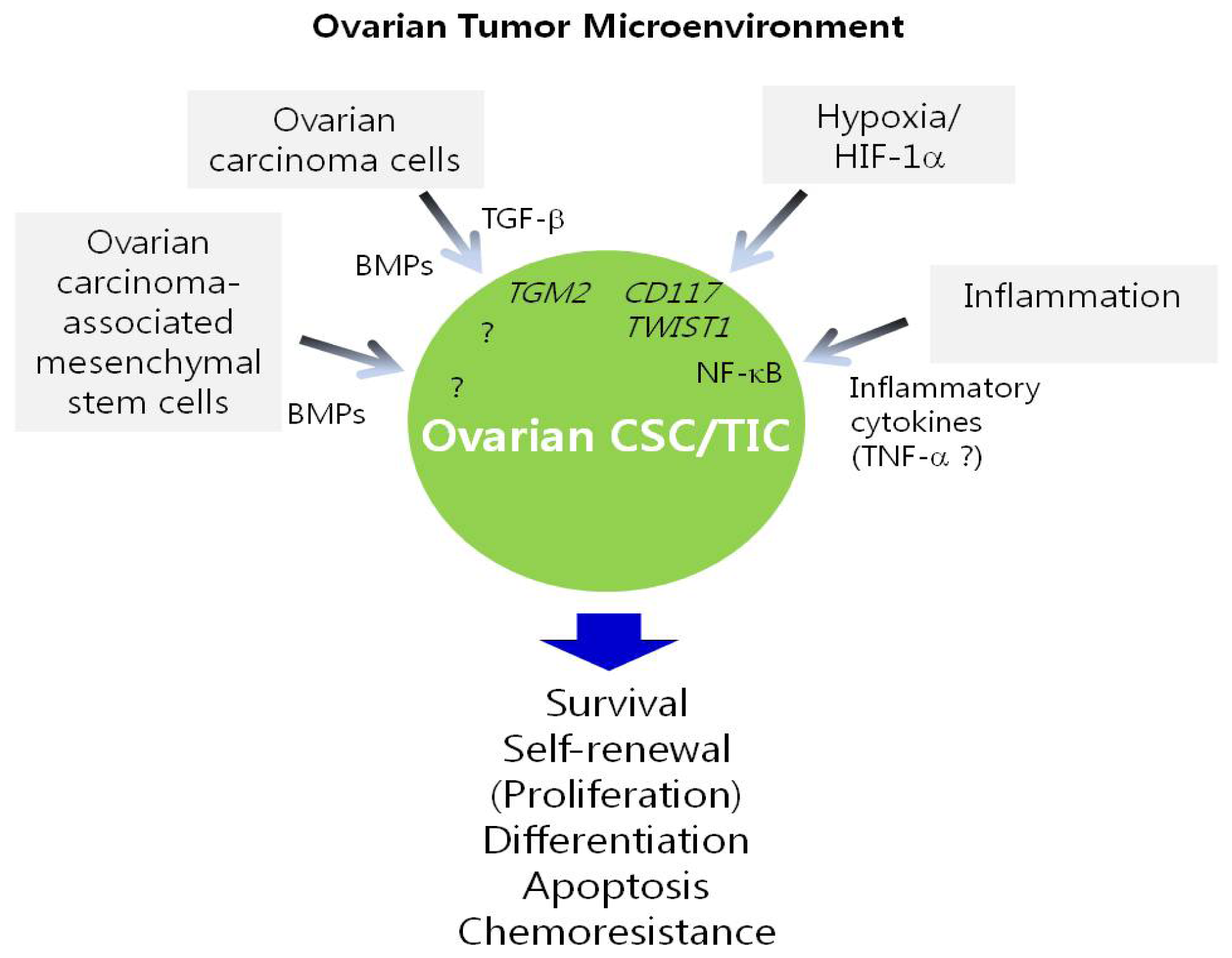
5. Regulation of Ovarian CSC/TICs by the Tumor Microenvironment
6. Genes and Pathways Involved in Chemoresistance and Recurrence of Ovarian Cancer
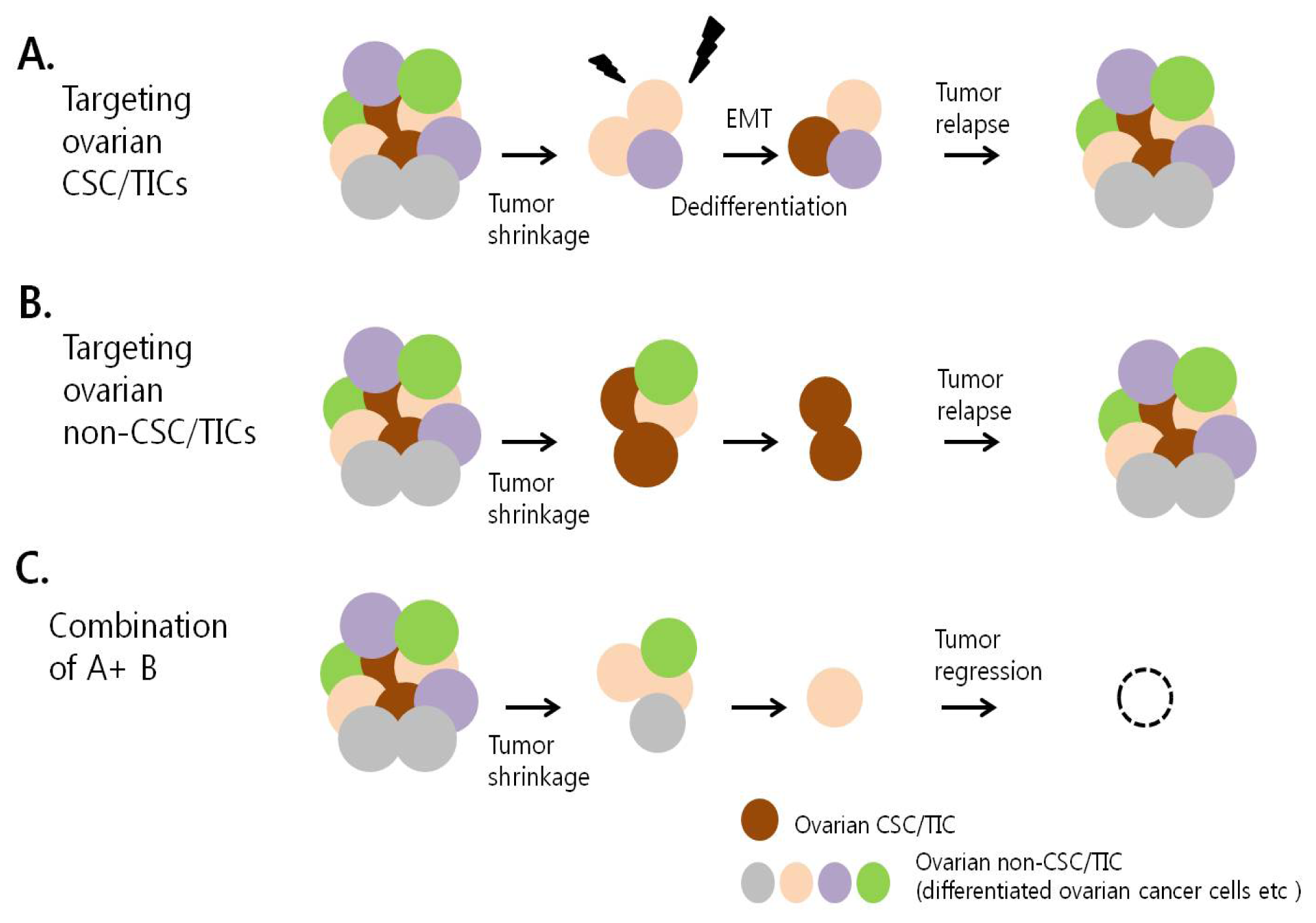
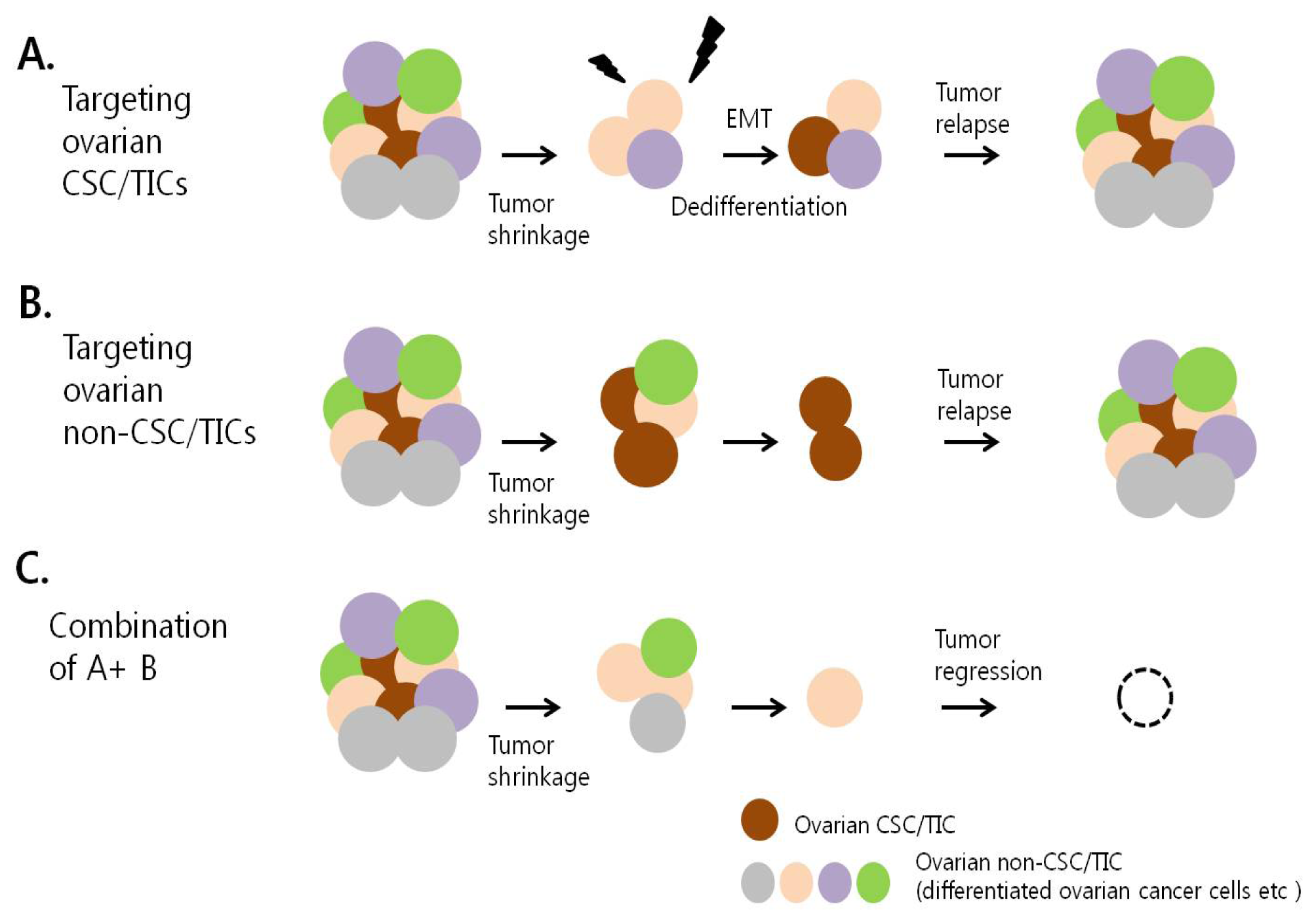
7. Therapeutic Targeting of Ovarian CSC/TICs
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA: A Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.[Green Version]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol 2012, 198, 281–293. [Google Scholar]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med 2011, 17, 313–319. [Google Scholar]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res 2005, 65, 3025–3029. [Google Scholar]
- Foster, R.; Buckanovich, R.J.; Rueda, B.R. Ovarian cancer stem cells: Working towards the root of stemness. Cancer Lett. 2012. [Google Scholar] [CrossRef]
- Wintzell, M.; Hjerpe, E.; Avall Lundqvist, E.; Shoshan, M. Protein markers of cancer-associated fibroblasts and tumor-initiating cells reveal subpopulations in freshly isolated ovarian cancer ascites. BMC Cancer 2012, 12, 359. [Google Scholar]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar]
- Moserle, L.; Indraccolo, S.; Ghisi, M.; Frasson, C.; Fortunato, E.; Canevari, S.; Miotti, S.; Tosello, V.; Zamarchi, R.; Corradin, A.; et al. The side population of ovarian cancer cells is a primary target of IFN-alpha antitumor effects. Cancer Res 2008, 68, 5658–5668. [Google Scholar]
- Hu, L.; McArthur, C.; Jaffe, R.B. Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br. J. Cancer 2010, 102, 1276–1283. [Google Scholar]
- Meng, E.; Long, B.; Sullivan, P.; McClellan, S.; Finan, M.A.; Reed, E.; Shevde, L.; Rocconi, R.P. CD44+/CD24− ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin. Exp. Metastasis 2012, 29, 939–948. [Google Scholar]
- Zhang, J.; Guo, X.; Chang, D.Y.; Rosen, D.G.; Mercado-Uribe, I.; Liu, J. CD133 expression associated with poor prognosis in ovarian cancer. Mod. Pathol 2012, 25, 456–464. [Google Scholar]
- Deng, S.; Yang, X.; Lassus, H.; Liang, S.; Kaur, S.; Ye, Q.; Li, C.; Wang, L.P.; Roby, K.F.; Orsulic, S.; et al. Distinct expression levels and patterns of stem cell marker, aldehyde dehydrogenase isoform 1 (ALDH1), in human epithelial cancers. PLoS One 2010, 5, e10277. [Google Scholar]
- Landen, C.N., Jr; Goodman, B.; Katre, A.A.; Steg, A.D.; Nick, A.M.; Stone, R.L.; Miller, L.D.; Mejia, P.V.; Jennings, N.B.; Gershenson, D.M.; et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther. 2010, 9, 3186–3199. [Google Scholar]
- Wang, Y.C.; Yo, Y.T.; Lee, H.Y.; Liao, Y.P.; Chao, T.K.; Su, P.H.; Lai, H.C. ALDH1-bright epithelial ovarian cancer cells are associated with CD44 expression, drug resistance, and poor clinical outcome. Am. J. Pathol 2012, 180, 1159–1169. [Google Scholar]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res 2011, 71, 3991–4001. [Google Scholar]
- Ricci, F.; Bernasconi, S.; Perego, P.; Ganzinelli, M.; Russo, G.; Bono, F.; Mangioni, C.; Fruscio, R.; Signorelli, M.; Broggini, M.; et al. Ovarian carcinoma tumor-initiating cells have a mesenchymal phenotype. Cell Cycle 2012, 11, 1966–1976. [Google Scholar]
- Wintzell, M.; Lofstedt, L.; Johansson, J.; Pedersen, A.B.; Fuxe, J.; Shoshan, M. Repeated cisplatin treatment can lead to a multiresistant tumor cell population with stem cell features and sensitivity to 3-bromopyruvate. Cancer Biol. Ther 2012, 13, 1454–1462. [Google Scholar]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68, 4311–4320. [Google Scholar]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar]
- Wei, X.; Dombkowski, D.; Meirelles, K.; Pieretti-Vanmarcke, R.; Szotek, P.P.; Chang, H.L.; Preffer, F.I.; Mueller, P.R.; Teixeira, J.; MacLaughlin, D.T.; et al. Mullerian inhibiting substance preferentially inhibits stem/progenitors in human ovarian cancer cell lines compared with chemotherapeutics. Proc. Natl. Acad. Sci. USA 2010, 107, 18874–18879. [Google Scholar]
- Luo, L.; Zeng, J.; Liang, B.; Zhao, Z.; Sun, L.; Cao, D.; Yang, J.; Shen, K. Ovarian cancer cells with the CD117 phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp. Mol. Pathol 2011, 91, 596–602. [Google Scholar]
- Gao, M.Q.; Choi, Y.P.; Kang, S.; Youn, J.H.; Cho, N.H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010, 29, 2672–2680. [Google Scholar]
- Shi, M.F.; Jiao, J.; Lu, W.G.; Ye, F.; Ma, D.; Dong, Q.G.; Xie, X. Identification of cancer stem cell-like cells from human epithelial ovarian carcinoma cell line. Cell. Mol. Life Sci 2010, 67, 3915–3925. [Google Scholar]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer. Journal International du Cancer 2012, 130, 29–39. [Google Scholar]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol 2011, 8, 97–106. [Google Scholar]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog--a cancer stem cell pathway. Clin. Cancer Res 2010, 16, 3130–3140. [Google Scholar]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar]
- Park, J.T.; Li, M.; Nakayama, K.; Mao, T.L.; Davidson, B.; Zhang, Z.; Kurman, R.J.; Eberhart, C.G.; Shih Ie, M.; Wang, T.L. Notch3 gene amplification in ovarian cancer. Cancer Res 2006, 66, 6312–6318. [Google Scholar]
- Park, J.T.; Chen, X.; Trope, C.G.; Davidson, B.; Shih Ie, M.; Wang, T.L. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am. J. Pathol 2010, 177, 1087–1094. [Google Scholar]
- Ivan, C.; Hu, W.; Bottsford-Miller, J.; Zand, B.; Dalton, H.J.; Liu, T.; Huang, J.; Nick, A.M.; Lopez-Berestein, G.; Coleman, R.L.; et al. Epigenetic analysis of the Notch superfamily in high-grade serous ovarian cancer. Gynecol. Oncol 2012, 128, 506–511. [Google Scholar]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.H.; et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar]
- Liao, X.; Siu, M.K.; Au, C.W.; Wong, E.S.; Chan, H.Y.; Ip, P.P.; Ngan, H.Y.; Cheung, A.N. Aberrant activation of hedgehog signaling pathway in ovarian cancers: effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar]
- Chen, X.; Horiuchi, A.; Kikuchi, N.; Osada, R.; Yoshida, J.; Shiozawa, T.; Konishi, I. Hedgehog signal pathway is activated in ovarian carcinomas, correlating with cell proliferation: it’s inhibition leads to growth suppression and apoptosis. Cancer Sci 2007, 98, 68–76. [Google Scholar]
- Bhattacharya, R.; Kwon, J.; Ali, B.; Wang, E.; Patra, S.; Shridhar, V.; Mukherjee, P. Role of hedgehog signaling in ovarian cancer. Clin. Cancer Res 2008, 14, 7659–7666. [Google Scholar]
- McCann, C.K.; Growdon, W.B.; Kulkarni-Datar, K.; Curley, M.D.; Friel, A.M.; Proctor, J.L.; Sheikh, H.; Deyneko, I.; Ferguson, J.A.; Vathipadiekal, V.; et al. Inhibition of Hedgehog signaling antagonizes serous ovarian cancer growth in a primary xenograft model. PLoS One 2011, 6, e28077. [Google Scholar]
- Ray, A.; Meng, E.; Reed, E.; Shevde, L.A.; Rocconi, R.P. Hedgehog signaling pathway regulates the growth of ovarian cancer spheroid forming cells. Int. J. Oncol 2011, 39, 797–804. [Google Scholar]
- Celia-Terrassa, T.; Meca-Cortes, O.; Mateo, F.; de Paz, A.M.; Rubio, N.; Arnal-Estape, A.; Ell, B.J.; Bermudo, R.; Diaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig 2012, 122, 1849–1868. [Google Scholar]
- Jiang, H.; Lin, X.; Liu, Y.; Gong, W.; Ma, X.; Yu, Y.; Xie, Y.; Sun, X.; Feng, Y.; Janzen, V.; et al. Transformation of Epithelial Ovarian Cancer Stemlike Cells into Mesenchymal Lineage via EMT Results in Cellular Heterogeneity and Supports Tumor Engraftment. Mol. Med 2012, 18, 1197–1208. [Google Scholar]
- Cao, L.; Shao, M.; Schilder, J.; Guise, T.; Mohammad, K.S.; Matei, D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar]
- Barbolina, M.V.; Burkhalter, R.J.; Stack, M.S. Diverse mechanisms for activation of Wnt signalling in the ovarian tumour microenvironment. Biochem. J 2011, 437, 1–12. [Google Scholar]
- Malanchi, I.; Peinado, H.; Kassen, D.; Hussenet, T.; Metzger, D.; Chambon, P.; Huber, M.; Hohl, D.; Cano, A.; Birchmeier, W.; et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 2008, 452, 650–653. [Google Scholar]
- Majeti, R.; Becker, M.W.; Tian, Q.; Lee, T.L.; Yan, X.; Liu, R.; Chiang, J.H.; Hood, L.; Clarke, M.F.; Weissman, I.L. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3396–3401. [Google Scholar]
- Leizer, A.L.; Alvero, A.B.; Fu, H.H.; Holmberg, J.C.; Cheng, Y.C.; Silasi, D.A.; Rutherford, T.; Mor, G. Regulation of inflammation by the NF-kappaB pathway in ovarian cancer stem cells. Am. J. Reprod. Immunol 2011, 65, 438–447. [Google Scholar]
- Chefetz, I.; Alvero, A.B.; Holmberg, J.C.; Lebowitz, N.; Craveiro, V.; Yang-Hartwich, Y.; Yin, G.; Squillace, L.; Gurrea Soteras, M.; Aldo, P.; et al. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle 2013, 12, 511–521. [Google Scholar]
- Bowtell, D.D. The genesis and evolution of high-grade serous ovarian cancer. Nat. Rev. Cancer 2010, 10, 803–808. [Google Scholar]
- Motohara, T.; Masuko, S.; Ishimoto, T.; Yae, T.; Onishi, N.; Muraguchi, T.; Hirao, A.; Matsuzaki, Y.; Tashiro, H.; Katabuchi, H.; et al. Transient depletion of p53 followed by transduction of c-Myc and K-Ras converts ovarian stem-like cells into tumor-initiating cells. Carcinogenesis 2011, 32, 1597–1606. [Google Scholar]
- Chau, W.K.; Ip, C.K.; Mak, A.S.; Lai, H.C.; Wong, A.S. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/beta-catenin-ATP-binding cassette G2 signaling. Oncogene 2012. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Zhang, W.Z.; Wang, L.H. Regulation of cancer cell survival, migration, and invasion by twist: AKT2 comes to interplay. Cancer Res 2008, 68, 957–960. [Google Scholar]
- Yin, G.; Chen, R.; Alvero, A.B.; Fu, H.H.; Holmberg, J.; Glackin, C.; Rutherford, T.; Mor, G. TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through MIR199A2/214. Oncogene 2010, 29, 3545–3553. [Google Scholar]
- Massard, C.; Deutsch, E.; Soria, J.C. Tumour stem cell-targeted treatment: elimination or differentiation. Ann. Oncol.: Off. J. Euro. Soc. Med. Oncol. /ESMO 2006, 17, 1620–1624. [Google Scholar]
- Yin, G.; Alvero, A.B.; Craveiro, V.; Holmberg, J.C.; Fu, H.H.; Montagna, M.K.; Yang, Y.; Chefetz-Menaker, I.; Nuti, S.; Rossi, M.; et al. Constitutive proteasomal degradation of TWIST-1 in epithelial-ovarian cancer stem cells impacts differentiation and metastatic potential. Oncogene 2012, 32, 39–49. [Google Scholar]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar]
- Lu, L.; Katsaros, D.; Mayne, S.T.; Risch, H.A.; Benedetto, C.; Canuto, E.M.; Yu, H. Functional study of risk loci of stem cell-associated gene lin-28B and associations with disease survival outcomes in epithelial ovarian cancer. Carcinogenesis 2012, 33, 2119–2125. [Google Scholar]
- Siu, M.K.; Wong, E.S.; Kong, D.S.; Chan, H.Y.; Jiang, L.; Wong, O.G.; Lam, E.W.; Chan, K.K.; Ngan, H.Y.; Le, X.F.; et al. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene 2012. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lin, X.; Zhong, X.; Kaur, S.; Li, N.; Liang, S.; Lassus, H.; Wang, L.; Katsaros, D.; Montone, K.; et al. Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1-positive cancer stem cells. Cancer Res 2010, 70, 9463–9472. [Google Scholar]
- Peng, S.; Maihle, N.J.; Huang, Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene 2010, 29, 2153–2159. [Google Scholar]
- Xu, C.X.; Xu, M.; Tan, L.; Yang, H.; Permuth-Wey, J.; Kruk, P.A.; Wenham, R.M.; Nicosia, S.V.; Lancaster, J.M.; Sellers, T.A.; et al. MicroRNA MiR-214 Regulates Ovarian Cancer Cell Stemness by Targeting p53/Nanog. J. Biol. Chem 2012, 287, 34970–34978. [Google Scholar]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.H.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet 2007, 39, 237–242. [Google Scholar]
- Chapman-Rothe, N.; Curry, E.; Zeller, C.; Liber, D.; Stronach, E.; Gabra, H.; Ghaem-Maghami, S.; Brown, R. Chromatin H3K27me3/H3K4me3 histone marks define gene sets in high-grade serous ovarian cancer that distinguish malignant, tumour-sustaining and chemo-resistant ovarian tumour cells. Oncogene 2012. [Google Scholar] [CrossRef]
- Ventura, A.; Jacks, T. MicroRNAs and Cancer: Short RNAs Go a Long Way. Cell 2009, 136, 586–591. [Google Scholar]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar]
- Yu, F.; Deng, H.; Yao, H.; Liu, Q.; Su, F.; Song, E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene 2010, 29, 4194–4204. [Google Scholar]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol 2008, 10, 593–601. [Google Scholar]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008, 22, 894–907. [Google Scholar]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res 2008, 68, 9125–9130. [Google Scholar]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res 2009, 69, 7569–7576. [Google Scholar]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nature Med 2011, 17, 211–215. [Google Scholar]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 2009, 4, e6816. [Google Scholar]
- Ji, Q.; Hao, X.; Meng, Y.; Zhang, M.; Desano, J.; Fan, D.; Xu, L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 2008, 8, 266. [Google Scholar]
- Ji, J.; Yamashita, T.; Budhu, A.; Forgues, M.; Jia, H.L.; Li, C.; Deng, C.; Wauthier, E.; Reid, L.M.; Ye, Q.H.; et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology 2009, 50, 472–480. [Google Scholar]
- Cheng, W.; Liu, T.; Wan, X.; Gao, Y.; Wang, H. MicroRNA-199a targets CD44 to suppress the tumorigenicity and multidrug resistance of ovarian cancer-initiating cells. FEBS J 2012, 279, 2047–2059. [Google Scholar]
- Wu, Q.; Guo, R.; Lin, M.; Zhou, B.; Wang, Y. MicroRNA-200a inhibits CD133/1+ ovarian cancer stem cells migration and invasion by targeting E-cadherin repressor ZEB2. Gynecol. Oncol 2011, 122, 149–154. [Google Scholar]
- Nam, E.J.; Lee, M.; Yim, G.W.; Kim, J.H.; Kim, S.; Kim, S.W.; Kim, Y.T. MicroRNA profiling of a CD133(+) spheroid-forming subpopulation of the OVCAR3 human ovarian cancer cell line. BMC Med. Genomics 2012, 5. [Google Scholar] [CrossRef]
- Helland, A.; Anglesio, M.S.; George, J.; Cowin, P.A.; Johnstone, C.N.; House, C.M.; Sheppard, K.E.; Etemadmoghadam, D.; Melnyk, N.; Rustgi, A.K.; et al. Deregulation of MYCN, LIN28B and LET7 in a Molecular Subtype of Aggressive High-Grade Serous Ovarian Cancers. PLoS One 2011, 6, e18064. [Google Scholar]
- Kwon, M.J.; Shin, Y.K. Epigenetic Regulation of Cancer-Associated Genes in Ovarian Cancer. Int. J. Mol. Sci 2011, 12, 983–1008. [Google Scholar]
- Cabarcas, S.M.; Mathews, L.A.; Farrar, W.L. The cancer stem cell niche-there goes the neighborhood? Int. J. Cancer 2011, 129, 2315–2327. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Liang, D.; Ma, Y.; Liu, J.; Trope, C.G.; Holm, R.; Nesland, J.M.; Suo, Z. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer 2012, 12, 201. [Google Scholar]
- McLean, K.; Gong, Y.; Choi, Y.; Deng, N.; Yang, K.; Bai, S.; Cabrera, L.; Keller, E.; McCauley, L.; Cho, K.R.; et al. Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Investig 2011, 121, 3206–3219. [Google Scholar]
- Ma, W.; Ma, J.; Xu, J.; Qiao, C.; Branscum, A.; Cardenas, A.; Baron, A.T.; Schwartz, P.; Maihle, N.J.; Huang, Y. Lin28 regulates BMP4 and functions with Oct4 to affect ovarian tumor microenvironment. Cell Cycle 2012, 12, 88–97. [Google Scholar]
- Liao, C.P.; Adisetiyo, H.; Liang, M.; Roy-Burman, P. Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer Res 2010, 70, 7294–7303. [Google Scholar]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs Reprogram Normal Fibroblasts into Cancer-Associated Fibroblasts in Ovarian Cancer. Cancer Discov 2012, 2, 1100–1108. [Google Scholar]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res.: An Off. J. Am. Assoc. Cancer Res 2012, 18, 869–881. [Google Scholar]
- Zhou, B.B.S.; Zhang, H.Y.; Damelin, M.; Geles, K.G.; Grindley, J.C.; Dirks, P.B. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov 2009, 8, 806–823. [Google Scholar]
- Ziebarth, A.; Nowsheen, S.; Steg, A.D.; Shah, M.M.; Dobbin, Z.C.; Han, H.D.; Lopez-Berestein, G.; Sood, A.K.; Conner, M.B.; Yang, E.S.; et al. Endoglin (CD105) contributes to platinum resistance and is a target for tumor-specific therapy in epithelial ovarian cancer. Clin. Cancer Res 2012, 19, 170–182. [Google Scholar]
- Slomiany, M.G.; Dai, L.; Tolliver, L.B.; Grass, G.D.; Zeng, Y.; Toole, B.P. Inhibition of Functional Hyaluronan-CD44 Interactions in CD133-positive Primary Human Ovarian Carcinoma Cells by Small Hyaluronan Oligosaccharides. Clin. Cancer Res 2009, 15, 7593–7601. [Google Scholar]
- Casagrande, F.; Cocco, E.; Bellone, S.; Richter, C.E.; Bellone, M.; Todeschini, P.; Siegel, E.; Varughese, J.; Arin-Silasi, D.; Azodi, M.; et al. Eradication of chemotherapy-resistant CD44+ human ovarian cancer stem cells in mice by intraperitoneal administration of Clostridium perfringens enterotoxin. Cancer 2011, 117, 5519–5528. [Google Scholar]
- Yo, Y.T.; Lin, Y.W.; Wang, Y.C.; Balch, C.; Huang, R.L.; Chan, M.W.; Sytwu, H.K.; Chen, C.K.; Chang, C.C.; Nephew, K.P.; et al. Growth inhibition of ovarian tumor-initiating cells by niclosamide. Mol. Cancer Ther 2012, 11, 1703–1712. [Google Scholar]
- Alvero, A.B.; Montagna, M.K.; Holmberg, J.C.; Craveiro, V.; Brown, D.; Mor, G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol. Cancer Ther 2011, 10, 1385–1393. [Google Scholar]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol. Oncol 2012, 127, 390–397. [Google Scholar]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar]
- Visnyei, K.; Onodera, H.; Damoiseaux, R.; Saigusa, K.; Petrosyan, S.; De Vries, D.; Ferrari, D.; Saxe, J.; Panosyan, E.H.; Masterman-Smith, M.; et al. A Molecular Screening Approach to Identify and Characterize Inhibitors of Glioblastoma Stem Cells. Mol. Cancer Ther 2011, 10, 1818–1828. [Google Scholar]
- Mezencev, R.; Wang, L.; McDonald, J.F. Identification of inhibitors of ovarian cancer stem-like cells by high-throughput screening. J. Ovarian Res 2012, 5, 30. [Google Scholar]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res 2009, 69, 7507–7511. [Google Scholar]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res 2011, 71, 3196–3201. [Google Scholar]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res. Treat 2011, 126, 355–364. [Google Scholar]
- Gotlieb, W.H.; Saumet, J.; Beauchamp, M.C.; Gu, J.; Lau, S.; Pollak, M.N.; Bruchim, I. In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol. Oncol 2008, 110, 246–250. [Google Scholar]
- Yasmeen, A.; Beauchamp, M.C.; Piura, E.; Segal, E.; Pollak, M.; Gotlieb, W.H. Induction of apoptosis by metformin in epithelial ovarian cancer: involvement of the Bcl-2 family proteins. Gynecol. Oncol 2011, 121, 492–498. [Google Scholar]
- Rattan, R.; Graham, R.P.; Maguire, J.L.; Giri, S.; Shridhar, V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 2011, 13, 483–491. [Google Scholar]
- Whitworth, J.M.; Londono-Joshi, A.I.; Sellers, J.C.; Oliver, P.J.; Muccio, D.D.; Atigadda, V.R.; Straughn, J.M., Jr; Buchsbaum, D.J. The impact of novel retinoids in combination with platinum chemotherapy on ovarian cancer stem cells. Gynecol. Oncol. 2012, 125, 226–230. [Google Scholar]

{kind=link}
{kind=link}
| Gene symbol | Gene name or target gene name | Types of ovarian CSC/TICs | Function | Reference |
|---|---|---|---|---|
| Genes or pathways (target gene) | ||||
| Notch signaling (NOTCH3) | Notch 3 | Side population | Expansion of ovarian CSC/TIC population Resistance to platinum | [36] |
| Hedgehog signaling | Growth of ovarian cancer spheroid-forming cells | [41] | ||
| TGF-β signaling (TGM2) | Transglutaminase 2 (C polypeptide, protein-glutamine-gamma-glut amyltransferase) | CD44+CD117+ cells | Self-renewal and expansion of ovarian CSC/TIC population | [44] |
| NF-κB signaling | CD44+ cells | Survival (apoptosis) | [48] | |
| TLR2-MyD88-NF-κB signaling | CD44+MyD88+ cells | Self-renewal | [49] | |
| KIT(c-kit, CD117) | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | Self-renewal and expansion of ovarian CSC/TICs Resistance to chemotherapy | [52] | |
| TP53 | p53 | EpCAM+ cells | Expansion of ovarian CSC/TIC population | [51] |
| LIN28 | Lin-28 homolog (C. elegans) | ALDH+ cells | Expansion of ovarian CSC/TIC population | [60] |
| TWIST1 | Twist basic helix-loop-helix transcription factor 1 | CD44+MyD88+ cells | Differentiation | [56] |
| miRNAs (target gene) | ||||
| miR-214 (TP53) | Tumor protein p53 | ALDH1+ cells | Self-renewal and expansion of ovarian CSC/TIC population | [62] |
| miR-199a (CD44) | CD44 molecule | CD44+CD117+ cells | Proliferation and invasion Resistance to chemotherapy | [78] |
| miR-200a (ZEB2) | Zinc finger E-box binding homeobox 2 | CD133+ cells | Migration and invasion | [79] |
| Agent or drug | Ovarian CSC/TICs targeted | Target | Effects | Mechanism of action | Reference |
|---|---|---|---|---|---|
| Targeting markers and stem cell pathways related to ovarian CSC/TICs | |||||
| Small hyaluronan oligosaccharides | Cells highly expressing CD133 | Hyaluronan-CD44 interaction | Inhibition of growth of ovarian carcinomas with high levels of CD133 in vivo | Inhibition (dissociation) of the hyaluronan-CD44 interaction | [93] |
| Clostridium perfringens enterotoxin (CPE) | CD44+ cells | Claudin-4 in CD44+ ovarian CSC/TICs | Inhibition of tumor progression of mice harboring xenografts of chemotherapy-resistant CD44+ ovarian CSC/TCS in vivo | CPE-induced cytotoxicity | [94] |
| γ-secretase inhibitors | Side population | Notch pathway | Increased sensitivity to cisplatin | Depletion of CSC/TICs | [36] |
| Targeting the function and properties of ovarian CSC/TICs | |||||
| Niclosamide | Side population | Metabolic pathways | Inhibition of ovarian CSC/TIC growth in vitro and in vivo | Disruption of multiple metabolic pathways | [95] |
| Isoflavane derivate, NV-128 | CD44+MyD88+ cells | Mitochondria (Mitochondrial bioenergetics) | Induction of apoptosis in ovarian CSC/TICs in vitro | Inhibition of mitochondrial function | [96] |
| Eriocalyxin B | CD44+ cells | NF-κB pathway | Induction of apoptosis in ovarian CSC/TICs in vitro | Inhibition of the NF-κB pathway | [48] |
| Metformin | ALDH+ cells | Inhibition of ovarian CSC/TIC growth in vitro and in vivo | Depletion of CSC/TICs, and inhibition of the formation of CSC/TIC spheres | [97] | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kwon, M.J.; Shin, Y.K. Regulation of Ovarian Cancer Stem Cells or Tumor-Initiating Cells. Int. J. Mol. Sci. 2013, 14, 6624-6648. https://doi.org/10.3390/ijms14046624
Kwon MJ, Shin YK. Regulation of Ovarian Cancer Stem Cells or Tumor-Initiating Cells. International Journal of Molecular Sciences. 2013; 14(4):6624-6648. https://doi.org/10.3390/ijms14046624
Chicago/Turabian StyleKwon, Mi Jeong, and Young Kee Shin. 2013. "Regulation of Ovarian Cancer Stem Cells or Tumor-Initiating Cells" International Journal of Molecular Sciences 14, no. 4: 6624-6648. https://doi.org/10.3390/ijms14046624