Distinct Signaling Cascades Elicited by Different Formyl Peptide Receptor 2 (FPR2) Agonists

Abstract
:1. Introduction
2. Microbe-Derived Peptides
3. Endogenous Peptides
3.1. Mitochondrial Peptides
3.2. Amyloidogenic Peptides and Proteins
3.3. Peptides Associated with Inflammatory and Anti-inflammatory Responses
3.4. Annexin A1 and Derived Peptides
3.5. Other Endogenous Peptides
4. Endogenous Nonpeptide Ligands
5. Ligands from Peptide Library
6. Ligands from Nonpeptide Library
Allergens
7. Conclusions
Acknowledgements
Conflict of Interest
References
- Chiang, N.; Serhan, C.N.; Dahlén, S.E.; Drazen, J.M.; Hay, D.W.; Rovati, G.E.; Shimizu, T.; Yokomizo, T.; Brink, C. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev 2006, 58, 463–487. [Google Scholar]
- Ying, G.; Iribarren, P.; Zhou, Y.; Gong, W.; Zhang, N.; Yu, Z.X.; Le, Y.; Cui, Y.; Wang, J.M. Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol 2004, 172, 7078–7085. [Google Scholar]
- He, R.; Sang, H.; Ye, R.D. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 2003, 101, 1572–1581. [Google Scholar]
- Le, Y.; Gong, W.; Tiffany, H.L.; Tumanov, A.; Nedospasov, S.; Shen, W.; Dunlop, N.M.; Gao, J.L.; Murphy, P.M.; Oppenheim, J.J.; et al. Amyloid β42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J. Neurosci. 2001, 21, RC123-1–RC123-5. [Google Scholar]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. USA 2002, 99, 1359–1364. [Google Scholar]
- Betten, A.; Bylund, J.; Cristophe, T.; Boulay, F.; Romero, A.; Hellstrand, K.; Dahlgren, C. A proinflammatory peptide from Helicobacter pylori activates monocytes to induce lymphocyte dysfunction and apoptosis. J. Clin. Invest 2001, 108, 1221–1228. [Google Scholar]
- Deng, X.; Ueda, H.; Su, S.B.; Gong, W.; Dunlop, N.M.; Gao, J.L.; Murphy, P.M.; Wang, J.M. A Synthetic Peptide Derived From Human Immunodeficiency Virus Type 1 gp120 Downregulates the Expression and Function of Chemokine Receptors CCR5 and CXCR4 in Monocytes by Activating the 7-Transmembrane G-Protein–Coupled Receptor FPRL1/LXA4R. Blood 1999, 94, 1165–1173. [Google Scholar]
- Rabiet, M.J.; Huet, E.; Boulay, F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur. J. Immunol 2005, 35, 2486–2495. [Google Scholar]
- de Paulis, A.; Prevete, N.; Rossi, F.W.; Rivellese, F.; Salerno, F.; Delfino, G.; Liccardo, B.; Avilla, E.; Montuori, N.; Mascolo, M.; et al. Helicobacter pylori Hp(2–20) Promotes Migration and Proliferation of Gastric Epithelial Cells by Interacting with Formyl Peptide Receptors In Vitro and Accelerates Gastric Mucosal Healing in vivo. J. Immunol 2009, 183, 3761–3769. [Google Scholar]
- Ali, H.; Richardson, R.M.; Haribabu, B.; Snyderman, R. Chemoattractant receptor cross-desensitization. J. Biol. Chem 1999, 274, 6027–6030. [Google Scholar]
- Shen, W.; Proost, P.; Li, B.; Gong, W.; Le, Y.; Sargeant, R.; Murphy, P.M.; Van Damme, J.; Wang, J.M. Activation of the Chemotactic Peptide Receptor FPRL1 in Monocytes Phosphorylates the Chemokine Receptor CCR5 and Attenuates Cell Responses to Selected Chemokines. Biochem. Biophys. Res. Comm 2000, 272, 276–283. [Google Scholar]
- Su, S.B.; Gao, J.L.; Gong, W.H.; Dunlop, N.M.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. T21/DP107, A Synthetic Leucine Zipper-Like Domain of the HIV-1 Envelope gp41, Attracts and Activates Human Phagocytes by Using G-Protein-Coupled Formyl Peptide Receptors. J. Immunol 1999, 162, 5924–5930. [Google Scholar]
- Le, Y.; Jiang, S.; Hu, J.; Gong, W.; Su, S.; Dunlop, N.M.; Shen, W.; Li, B.; Wang, J.M. N36, a Synthetic N-Terminal Heptad Repeat Domain of the HIV-1 Envelope Protein gp41, Is an Activator of Human Phagocytes. Clin. Immunol 2000, 96, 236–242. [Google Scholar]
- Lin, C.; Wei, W.; Zhang, J.; Liu, S.; Liu, Y.; Zheng, D. Formyl peptide receptor- like 1 mediated endogenous TRAIL gene expression with tumoricidal activity. Mol. Cancer Ther 2007, 6, 2618–2625. [Google Scholar]
- Kretschmer, D.; Gleske, A.K.; Rautenberg, M.; Wang, R.; Koberle, M.; Bohn, E.; Schoneberg, T.; Rabiet, M.J.; Boulay, F.; Klebanoff, S.J.; et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 2010, 7, 463–473. [Google Scholar]
- Forsman, H.; Christenson, K.; Bylund, J.; Dahlgren, C. Receptor-dependent and -independent immunomodulatory effects of phenol-soluble modulin peptides from Staphylococcus aureus on human neutrophils are abrogated through peptide inactivation by reactive oxygen species. Infect. Immun 2012, 80, 1987–1995. [Google Scholar]
- Chiang, N.; Fierro, I.M.; Gronert, K.; Serhan, C.N. Activation of Lipoxin A4 Receptors by Aspirin-triggered Lipoxins and Select Peptides Evokes Ligand-specific Responses in Inflammation. J. Exp. Med 2000, 191, 1197–1207. [Google Scholar]
- Seki, T.; Fukamizu, A.; Kiso, Y.; Mukai, H. Mitocryptide-2, a neutrophil-activating cryptide, is a specific endogenous agonist for formyl-peptide receptor-like 1. Biochem. Biophys. Res. Comm 2011, 404, 482–487. [Google Scholar]
- Stone, M.J. Amyloidosis: A final common pathway for protein deposition in tissues. Blood 1990, 75, 531–545. [Google Scholar]
- Su, S.B.; Gong, W.; Gao, J.L.; Shen, W.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. A Seven-transmembrane, G Protein–coupled Receptor, FPRL1, Mediates the Chemotactic Activityof Serum Amyloid A for Human Phagocytic Cells. J. Exp. Med 1999, 189, 395–402. [Google Scholar]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Lee, S.Y.; Lee, H.; Cho, K.H.; Yun, J.; Bae, Y.S. Serum Amyloid A Induces CCL2 Production via Formyl Peptide Receptor-Like 1-Mediated Signaling in Human Monocytes. J. Immunol 2008, 181, 4332–4339. [Google Scholar]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Kim, H.J.; Yun, J.; Baek, S.H.; Kim, K.; Bae, Y.S. A pertussis toxin sensitive G-protein-independent pathway is involved in serum amyloid A-induced formyl peptide receptor 2-mediated CCL2 production. Exp. Mol. Med 2010, 42, 302–309. [Google Scholar]
- Lee, H.Y.; Kim, M.K.; Park, K.S.; Shin, E.H.; Jo, S.H.; Kim, S.D.; Jo, E.J.; Lee, Y.N.; Lee, C.; Baek, S.H.; et al. Serum amyloid A induces contrary immune responses via formyl peptide receptor-like 1 in human monocytes. Mol. Pharmacol 2006, 70, 241–248. [Google Scholar]
- Lee, H.Y.; Kim, M.K.; Park, K.S.; Bae, Y.H.; Yun, J.; Park, J.I.; Kwak, J.Y.; Bae, Y.S. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem. Biophys. Res. Commun 2005, 330, 989–998. [Google Scholar]
- Lee, M.S.; Yoo, S.A.; Cho, C.S.; Suh, P.G.; Kim, W.U.; Ryu, S.H. Serum Amyloid A Binding to Formyl Peptide Receptor-Like 1 Induces Synovial Hyperplasia and Angiogenesis. J. Immunol 2006, 177, 5585–5594. [Google Scholar]
- O’Hara, R.; Murphy, E.P.; Whitehead, A.S.; FitzGerald, O.; Bresnihan, B. Local expression of the serum amyloid A and formyl peptide receptor-like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis. Arthritis Rheum 2004, 50, 1788–1799. [Google Scholar]
- Koga, T.; Torigoshi, T.; Motokawa, S.; Miyashita, T.; Maeda, Y.; Nakamura, M.; Komori, A.; Aiba, Y.; Uemura, T.; Yatsuhashi, H.H.; et al. Serum amyloid A-induced IL-6 production by rheumatoid synoviocytes. FEBS Lett. 2008, 582, 579–585. [Google Scholar]
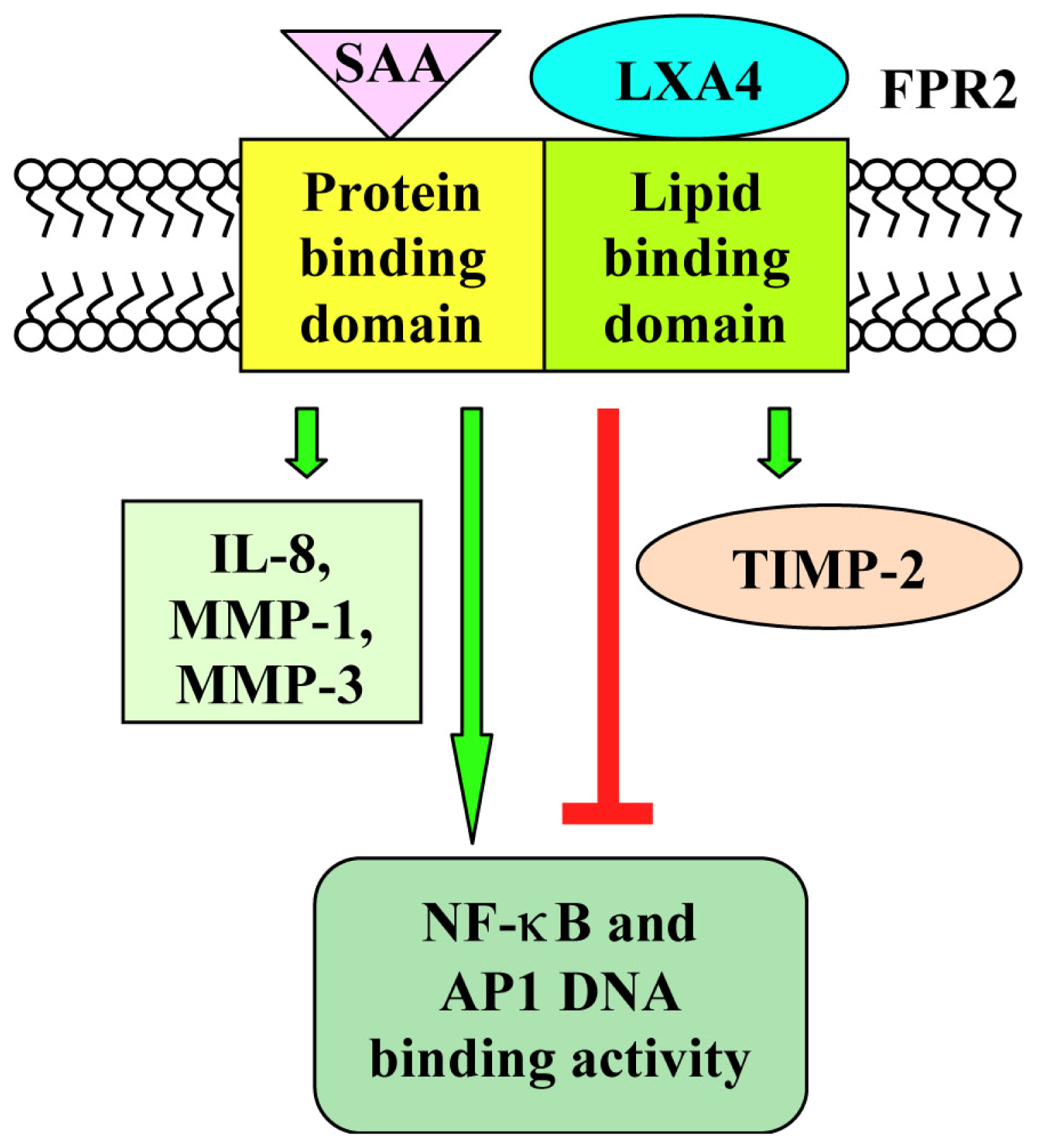
- Sodin-Semrl, S.; Spagnolo, A.; Mikus, R.; Barbaro, R.; Varga, J.; Fiore, S. Opposing regulation of interleukin-8 and Nf-kappaB responses by lipoxin A4 and serum amyloid A via the common lipoxin A receptor. Int. J. Immunopathol. Pharmacol 2004, 17, 145–156. [Google Scholar]
- Bozinovskia, S.; Uddin, M.; Vlahos, R.; Thompson, M.; McQualter, J.L.; Merritt, A.S.; Wark, P.A.; Hutchinson, A.; Irving, L.B.; Levy, B.D.; et al. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA 2012, 109, 935–940. [Google Scholar]
- Dong, Z.; An, F.; Wu, T.; Zhang, C.; Zhang, M.; Zhang, Y.; An, G.; An, F. PTX3, a Key Component of Innate Immunity, Is Induced by SAA via FPRL1-Mediated Signaling in HAECs. J. Cell. Biochem 2011, 112, 2097–2105. [Google Scholar]
- Baranova, I.N.; Vishnyakova, T.G.; Bocharov, A.V.; Kurlander, R.; Chen, Z.; Kimelman, M.L.; Remaley, A.T.; Csako, G.; Thomas, F.; Eggerman, T.L.; et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J. Biol. Chem 2005, 280, 8031–8040. [Google Scholar]
- Cheng, N.; He, R.; Tian, J.; Ye, P.P.; Ye, R.D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol 2008, 181, 22–26. [Google Scholar]
- Sandri, S.; Rodriguez, D.; Gomes, E.; Monteiro, H.P.; Russo, M.; Campa, A. Is serum amyloid A an endogenous TLR4 agonist? J. Leukoc. Biol 2008, 83, 1174–1180. [Google Scholar]
- Le, Y.; Ye, R.D.; Gong, W.; Li, J.; Iribarren, P.; Wang, J.M. Identification of functional domains in the formyl peptide receptor-like 1 for agonist-induced cell chemotaxis. FEBS J 2005, 272, 769–778. [Google Scholar]
- Brandeburg, L.O.; Konrad, M.; Wruck, C.; Koch, T.; Pufe, T.; Lucius, R. Involvement of formyl-peptide-receptor-like-1 and phospholipase D in the internalization and signal transduction of amyloid beta 1–42 in glial cells. Neuroscience 2008, 156, 266–276. [Google Scholar]
- Tiffany, H.L.; Lavigne, M.C.; Cui, Y.H.; Wang, J.M.; Leto, T.L.; Gao, J.L.; Murphy, P.M. Amyloid-β Induces Chemotaxis and Oxidant Stress by Acting at Formylpeptide Receptor 2, a G Protein-coupled Receptor Expressed in Phagocytes and Brain. J. Biol. Chem 2001, 276, 23645–23652. [Google Scholar]
- Brandeburg, L.O.; Konrad, M.; Wruck, C.; Koch, T.; Lucius, R.; Pufe, T. Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid beta 1–42-induced signal transduction in glial cells. J. Neurochem 2010, 113, 749–760. [Google Scholar]
- Harada, M.; Habata, Y.; Hosoya, M.; Nishi, K.; Fujii, R.; Kobayashi, M.; Hinuma, S. N-Formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochem. Biophys. Res. Commun 2004, 324, 255–261. [Google Scholar]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar]
- Hashimoto, Y.; Niikura, T.; Ito, Y.; Sudo, H.; Hata, M.; Arakawa, E.; Abe, Y.; Kita, Y.; Nishimoto, I. Detailed Characterization of Neuroprotection by a Rescue Factor Humanin against Various Alzheimer’s Disease-Relevant Insults. J. Neurosci 2001, 21, 9235–9245. [Google Scholar]
- Hashimoto, Y.; Suzuki, H.; Aiso, S.; Niikura, T.; Nishimoto, I.; Matsuoka, M. Involvement of tyrosine kinases and STAT3 in Humanin-mediated neuroprotection. Life Sci 2005, 77, 3092–3104. [Google Scholar]
- Le, Y.; Yazawa, H.; Gong, W.; Yu, Z.; Ferrans, V.J.; Murphy, P.M.; Wang, J.M. The Neurotoxic Prion Peptide Fragment PrP106–126 Is a Chemotactic Agonist for the G Protein-Coupled Receptor Formyl Peptide Receptor-Like 1. J. Immunol 2001, 166, 1448–1451. [Google Scholar]
- Zhoua, H.; Zhou, X.; Kouadir, M.; Zhang, Z.; Yin, X.; Yang, L.; Zhao, D. Induction of macrophage migration by neurotoxic prion protein fragment. J. Neurosci. Methods 2009, 181, 1–5. [Google Scholar]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and Signaling of Formyl-Peptide Receptors in the Brain. Neurochem. Res 2010, 35, 2018–2026. [Google Scholar]
- Brandenburg, L.O.; Koch, T.; Sievers, J.; Lucius, R. Internalization of PrP106–126 by the formyl-peptide-receptor-like-1 in glial cells. J. Neurochem 2007, 101, 718–728. [Google Scholar]
- Gargiulo, L.; Longanesi-Cattani, I.; Bifulco, K.; Franco, P.; Raiola, R.; Campiglia, P.; Grieco, P.; Peluso, G.; Stoppelli, M.P.; Carriero, M.V. Cross-talk beteween fMLP and vitronectin receptor triggered by urokinase receptor-derived SRSRY peptide. J.Biol. Chem 2005, 280, 25225–25232. [Google Scholar]
- de Paulis, A.; Montuori, N.; Prevete, N.; Fiorentino, I.; Rossi, F.W.; Visconte, V.; Rossi, G.; Marone, G.; Ragno, P. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J. Immunol 2004, 173, 5739–5748. [Google Scholar]
- Furlan, F.; Orlando, S.; Laudanna, C.; Resnati, M.; Basso, V.; Blasi, F.; Mondino, A. The soluble D2D3(88–274) fragment of the urokinase receptor inhibitsmonocyte chemotaxis and integrin-dependent cell adhesion. J. Cell Sci 2004, 117, 2909–2916. [Google Scholar]
- Jo, M.; Thomas, K.S.; Marozkina, N.; Amin, T.J.; Silva, C.M.; Parsons, S.J.; Gonias, S.L. Dynamic Assembly of the Urokinase-type Plasminogen Activator Signaling Receptor Complex Determines the Mitogenic Activity of Urokinase-type Plasminogen Activator. J. Biol. Chem 2005, 280, 17449–17457. [Google Scholar]
- Mazzieri, R.; D’Alessio, S.; Kenmoe, R.K.; Ossowski, L.; Blasi, F. An Uncleavable uPAR Mutant Allows Dissection of Signaling Pathways in uPA-dependent Cell Migration. Mol. Biol. Cell 2006, 17, 367–378. [Google Scholar]
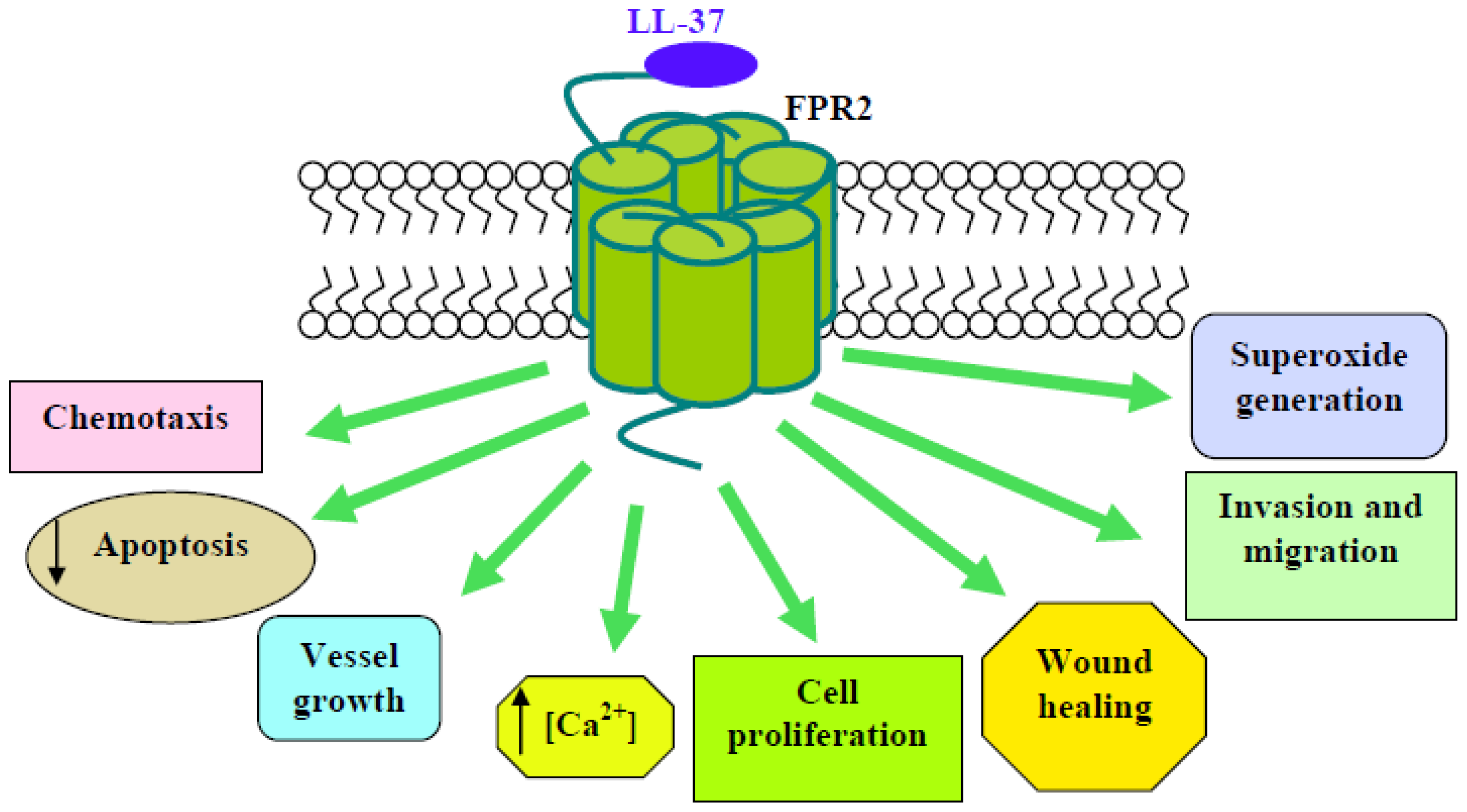
- Yang, D.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. LL-37, the Neutrophil Granule– and Epithelial cell–derived Cathelicidin, Utilizes Formyl Peptide Receptor–like 1 (FPRL1) as a Receptor to Chemoattract Human Peripheral Blood Neutrophils, Monocytes, and T Cells. J. Exp. Med. 2000, 192, 1069–1074. [Google Scholar]
- Nagaoka, I.; Tamura, H.; Hirata, M. An Antimicrobial Cathelicidin Peptide, Human CAP18/LL-37, Suppresses Neutrophil Apoptosis via the Activation of Formyl-Peptide Receptor-Like 1 and P2X7. J. Immunol 2006, 176, 3044–3052. [Google Scholar]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Lee, S.Y.; Yun, J.; Bae, Y.S. LL-37 inhibits serum amyloid A-induced IL-8 production in human neutrophils. Exp. Mol. Med 2009, 41, 325–333. [Google Scholar]
- Koczulla, R.; von Degenfeld, G.; Kupatt, C.; Krötz, F.; Zahler, S.; Gloe, T.; Issbrücker, K.; Unterberger, P.; Zaiou, M.; Lebherz, C.; et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J. Clin. Invest 2003, 111, 665–1672. [Google Scholar]
- Shaykhiev, R.; Beißwenger, C.; Kandler, K.; Senske, J.; Puchner, A.; Damm, T.; Behr, J.; Bals, R. Human endogenous antibiotic LL-37 stimulates airway epithelial cell proliferation and wound closure. Am. J. Physiol. Lung Cell. Mol. Physiol 2005, 289, L842–L848. [Google Scholar]
- Coffelt, S.B.; Marini, F.C.; Watson, K.; Zwezdaryk, K.J.; Dembinski, J.L.; LaMarca, H.L.; Tomchuck, S.L.; Honer zu Bentrup, K.; Danka, E.S.; Henkle, S.L.; et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3806–3811. [Google Scholar]
- Coffelt, S.B.; Tomchuck, S.L.; Zwezdaryk, K.J.; Danka, E.S.; Scandurro, A.B. Leucine Leucine-37 Uses Formyl Peptide Receptor–Like 1 to Activate Signal Transduction Pathways, Stimulate Oncogenic Gene Expression, and Enhance the Invasiveness of Ovarian Cancer Cells. Mol. Cancer Res 2009, 7, 907–915. [Google Scholar]
- Li, Y.; Cai, L.; Wang, H.; Wu, P.; Gu, W.; Chen, Y.; Hao, H.; Tang, K.; Yi, P.; Liu, M.; et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene 2011, 30, 3887–3899. [Google Scholar]
- Wan, M.; Godson, C.; Guiry, P.J.; Agerberth, B.; Haeggström, J.Z. Leukotriene B4/antimicrobial peptide LL-37 proinflammatory circuits are mediated by BLT1 and FPR2/ALX and are counterregulated by lipoxin A4 and resolving E1. FASEB J 2011, 25, 1697–1705. [Google Scholar]
- Gaudreault, E.; Gosselin, J. Leukotriene B4 induces release of antimicrobial peptides in lungs of virtally infected mice. J. Immunol 2008, 180, 6211–6221. [Google Scholar]
- Iaccio, A.; Cattaneo, F.; Mauro, M.; Ammendola, R. FPRL1-mediated induction of superoxide in LL-37-stimulated IMR90 human fibroblasts. Arch. Biochem. Biophys 2009, 481, 94–100. [Google Scholar]
- Elagoz, A.; Henderson, D.; Babu, P.S.; Salter, S.; Grahames, C.; Bowers, L.; Roy, M.O.; Laplante, P.; Grazzini, E.; Ahmad, S.; et al. A truncated form of CKβ8–1 is a potent agonist for human formyl peptide-receptor-like 1 receptor. Br. J. Pharmacol 2004, 141, 37–46. [Google Scholar]
- Miao, Z.; Premack, B.A.; Wei, Z.; Wang, Y.; Gerard, C.; Showell, H.; Howard, M.; Schall, T.J.; Berahovich, R. Proinflammatory Proteases Liberate a Discrete High-Affinity Functional FPRL1 (CCR12) Ligand from CCL23. J. Immunol 2007, 178, 7395–7404. [Google Scholar]
- Vaudry, D.; Gonzalez, B.J.; Basille, M.; Yon, L.; Fournier, A.; Vaudry, H. Pituitary adenylate cyclase-activating polypeptide and its receptors: from structure to functions. Pharmacol. Rev 2000, 52, 269–324. [Google Scholar]
- Delgado, M.; Pozo, D.; Ganea, D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol. Rev 2004, 56, 249–290. [Google Scholar]
- Chorny, A.; Gonzalez-Rey, E.; Varela, N.; Robledo, G.; Delgado, M. Signaling mechanisms of vasoactive intestinal peptide in inflammatory conditions. Regul. Pept 2006, 137, 67–74. [Google Scholar]
- El Zein, N.; Badran, B.; Sariban, E. VIP differentially activates β2 integrins, CR1, and matrix metalloproteinase-9 in human monocytes through cAMP/PKA, EPAC, and PI-3K signaling pathways via VIP receptor type 1 and FPRL1. J. Leukoc. Biol 2008, 83, 972–981. [Google Scholar]
- Hoyle, C.H. Neuropeptide families: evolutionary perspectives. Regul. Pept 1998, 73, 1–33. [Google Scholar]
- Kim, Y.; Lee, B.D.; Kim, O.; Bae, Y.S.; Lee, T.; Suh, P.G.; Ryu, S.H. Pituitary Adenylate Cyclase-Activating Polypeptide 27 Is a Functional Ligand for Formyl Peptide Receptor-Like 1. J. Immunol 2006, 176, 2969–2975. [Google Scholar]
- El Zein, N.; Badran, B.; Sariban, E. The neuropeptide pituitary adenylate cyclase activating polypeptide modulates Ca2+ and pro-inflammatory functions in human monocytes through the G protein-coupled receptors VPAC-1 and formyl peptide receptor-like 1. Cell Calcium 2008, 43, 270–284. [Google Scholar]
- Ernst, S.; Lange, C.; Wilbers, A.; Goebeler, V.; Gerke, V.; Rescher, U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J. Immunol 2004, 172, 7669–7676. [Google Scholar]
- Gavins, F.N.; Yona, S.; Kamal, A.M.; Flower, R.J.; Perretti, M. Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood 2003, 101, 4140–4147. [Google Scholar]
- Hayhoe, R.P.; Kamal, A.M.; Solito, E.; Flower, R.J.; Cooper, D.; Perretti, M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow- indication of distinct receptor involvement. Blood 2006, 107, 2123–2130. [Google Scholar]
- Solito, E.; Romero, I.A.; Marullo, S.; Russo-Marie, F.; Weksler, B.B. Annexin 1 binds to U937 monocytic cells and inhibits their adhesion to microvascular endothelium: involvement of the alpha 4 beta 1 integrin. J. Immunol 2000, 165, 1573–1581. [Google Scholar]
- Perretti, M.; Chiang, N.; La, M.; Fierro, I.M.; Marullo, S.; Getting, S.J.; Solito, E.; Serhan, C.N. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med 2002, 8, 1296–1302. [Google Scholar]
- Kamal, A.M.; Hayhoe, R.P.; Paramasivam, A.; Cooper, D.; Flower, R.J.; Solito, E.; Perretti, M. Antiflammin-2 activates the human formyl-peptide receptor like 1. Sci. World J 2006, 6, 1375–1384. [Google Scholar]
- Movitz, C.; Brive, L.; Hellstrand, K.; Rabiet, M.J.; Dahlgren, C. The Annexin I Sequence Gln9-Ala10-Trp11-Phe12 Is a Core Structure for Interaction with the Formyl Peptide Receptor 1. J. Biol. Chem 2010, 19, 14338–14345. [Google Scholar]
- Tagoe, C.E.; Marjanovic, N.; Park, J.Y.; Chan, E.S.; Abeles, A.M.; Attur, M.; Abramson, S.B.; Pillinger, M.H. Metalloproteinase Secretion from Rheumatoid Arthritis Synovial Fibroblasts. J. Immunol 2008, 181, 2813–2820. [Google Scholar]
- Ang, E.Z.; Nguyen, H.T.; Sim, H.L.; Putti, T.C.; Lim, L.H. Annexin-1 regulates growth arrest induced by high levels of estrogen in MCF-7 breast cancer cells. Mol. Cancer Res 2009, 7, 266–274. [Google Scholar]
- Babbin, B.A.; Lee, W.Y.; Parkos, C.A.; Winfree, L.M.; Akyildiz, A.; Perretti, M.; Nusrat, A. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J. Biol. Chem 2006, 281, 19588–19599. [Google Scholar]
- Khau, T.; Langenbach, S.Y.; Schuliga, M.; Harris, T.; Johnstone, C.N.; Anderson, R.L.; Stewart, A.G. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. Regulation of breast cancer cell proliferation. FASEB J 2011, 25, 483–496. [Google Scholar]
- Jia, Y.; Morand, E.F.; Song, W.; Cheng, Q.; Stewart, A.; Yang, Y.H. Regulation of lung fibroblasts activation by annexin 1. J. Cell. Physiol 2013, 228, 476–484. [Google Scholar]
- Bena, S.; Brancaleone, V.; Wang, J.M.; Perretti, M.; Flower, R.J. Annexin A1 Interaction with the FPR2/ALX Receptor: identification of distinct domains and downstrean associated signaling. J. Biol. Chem 2012, 287, 24690–24697. [Google Scholar]
- Kirpotina, L.N.; Khlebnikov, A.I.; Schepetkin, I.A.; Ye, R.D.; Rabiet, M.J.; Jutila, M.A.; Quinn, M.T. Identification of novel small molecule agonists for human formyl peptide receptors and pharmacophore models of their recognition. Mol. Pharmacol 2010, 77, 159–170. [Google Scholar]
- Chen, Q.; Wade, D.; Kurosaka, K.; Wang, Z.Y.; Oppenheim, J.J.; Yang, D. Temporin A and related frog antimicrobial peptides use formyl peptide receptorlike 1 as a receptor to chemoattract phagocytes. J. Immunol 2004, 173, 2652–2659. [Google Scholar]
- Madenspacher, J.H.; Azzam, K.M.; Gong, W.; Gowdy, K.M.; Vitek, M.P.; Laskowitz, D.T.; Remaley, A.T.; Wang, J.M.; Fessler, M.B. Apolipoproteins and apolipoprotein mimetic peptides modulate phagocyte trafficking through chemotactic activity. J. Biol. Chem 2012, 287, 43730–43740. [Google Scholar]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 141–162. [Google Scholar]
- Fiore, S.; Maddox, J.F.; Perez, H.D.; Serhan, C.N. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med 1994, 180, 253–260. [Google Scholar]
- Fiore, S.; Serhan, C.N. Lipoxin A4 Receptor Activation Is Distinct from That of the Formyl Peptide Receptor in Myeloid Cells: Inhibition of CD 11/18 Expression by Lipoxin A4-Lipoxin A4 Receptor Interaction. Biochemistry 1995, 34, 16678–16686. [Google Scholar]
- Murphy, P.M.; Ozcelik, T.; Kenney, R.T.; Tiffany, H.L.; McDermott, D.; Francke, U. A structural homologue of the N-formyl peptide receptor. Characterization and chromosome mapping of a peptide chemoattractant receptor family. J. Biol. Chem 1992, 267, 7637–7643. [Google Scholar]
- Perez, H.D.; Holmes, R.; Kelly, E.; McClary, J.; Andrews, W.H. Cloning of a cDNA encoding a receptor related to the formyl peptide receptor of human neutrophils. Gene 1992, 118, 303–304. [Google Scholar]
- Ye, R.D.; Cavanagh, S.L.; Quehenberger, O.; Prossnitz, E.R.; Cochrane, C.G. Isolation of a cDNA that encodes a novel granulocyte N-formyl peptide receptor. Biochem. Biophys. Res. Commun 1992, 184, 582–589. [Google Scholar]
- Colgan, S.P.; Serhan, C.N.; Parkos, C.A.; Delp-Archer, C.; Madara, J.L. Lipoxin A4 Modulates Transmigration of Human Neutrophils across Intestinal Epithelial Monolayers. J. Clin. Invest 1993, 92, 75–82. [Google Scholar]
- Kucharzik, T.; Gewirtz, A.T.; Merlin, D.; Madara, J.L.; Williams, I.R. Lateral membrane LXA4 receptors mediate LXA4’s anti-inflammatory actions on intestinal epithelium. Am. J. Physiol. Cell Physiol 2003, 284, C888, -C-896.. [Google Scholar]
- Paul-Clark, M.J.; Van Cao, T.; Moradi-Bidhendi, N.; Cooper, D.; Gilroy, D.W. 15-epi-lipoxin A4-mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J. Exp. Med 2004, 200, 69–78. [Google Scholar]
- Bae, Y.S.; Park, J.C.; He, R.; Ye, R.D.; Kwak, J.Y.; Suh, P.G.; Ryu, S.H. Differential Signaling of Formyl Peptide Receptor-Like 1 by Trp-Lys-Tyr-Met-Val-Met-CONH2 or Lipoxin A4 in Human Neutrophils. Mol. Pharmacol 2003, 64, 721–730. [Google Scholar]
- McMahon, B.; Stenson, C.; McPhillips, F.; Fanning, A.; Brady, H.R.; Godson, C. Lipoxin A4 antagonizes the mitogenic effects of leukotriene D4 in human renal mesengial cells. J. Biol. Chem 2000, 275, 27566–27575. [Google Scholar]
- Mitchell, D.; Rodgers, K.; Hanly, J.; McMahon, B.; Brady, H.R.; Martin, F.; Godson, C. Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am. J. Pathol 2004, 164, 937–946. [Google Scholar]
- Wu, S.H.; Wu, X.H.; Lu, C.; Dong, L.; Chen, Z.Q. Lipoxin A4 inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am. J. Respir. Cell. Mol. Biol 2006, 34, 65–72. [Google Scholar]
- Sodin-Semrl, S.; Taddeo, B.; Tseng, D.; Varga, J.; Fiore, S. Lipoxin A4 inhibits IL1β-induced IL-6, IL-8 and matrix metalloproteinase-3 production in human synovial fibroblasts and enhances synthesis of tissue inhibitors of metalloproteinases. J. Immunol 2000, 164, 2660–2666. [Google Scholar]
- Fiore, S.; Antico, G.; Aloman, M.; Sodin-Semrl, S. Lipoxin A4 biology in the human synovium. Role of the ALX signaling pathways in modulation of inflammatory arthritis. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 189–196. [Google Scholar]
- Souza, D.G.; Fagundes, C.T.; Amaral, F.A.; Cisalpino, D.; Sousa, L.P.; Vieira, A.T.; Pinho, V.; Nicoli, J.R.; Vieira, L.Q.; Fierro, I.M.; et al. The Required Role of Endogenously Produced Lipoxin A4 and Annexin-1 for the Production of IL-10 and Inflammatory Hyporesponsiveness in Mice. J. Immunol 2007, 179, 8533–8543. [Google Scholar]
- Nascimento-Silva, V.; Arruda, M.A.; Barja-Fidalgo, C.; Villela, C.G.; Fierro, I.M. Novel lipid mediator aspirin-triggered lipoxin A 4 induces heme oxygenase-1 in endothelial cells. Am. J. Physiol. Cell. Physiol 2005, 289, C557–C563. [Google Scholar]
- Bonnans, C.; Gras, D.; Chavis, C.; Mainprice, B.; Vachier, I.; Godard, P.; Chanez, P. Synthesis and anti-inflammatory effect of lipoxins in human airway epithelial cells. Biomed. Pharmacother 2007, 61, 261–267. [Google Scholar]
- Machado, F.S.; Esper, L.; Dias, A.; Madan, R.; Gu, Y.; Hildeman, D.; Serhan, C.N.; Karp, C.L.; Aliberti, J. Native and aspirin-triggered lipoxins control innate immunity by inducing proteasomal degradation of TRAF6. J. Exp. Med 2008, 205, 1077–1086. [Google Scholar]
- Machado, F.S.; Johndrow, J.E.; Esper, L.; Dias, A.; Bafica, A.; Serhan, C.N.; Aliberti, J. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat. Med 2006, 12, 330–334. [Google Scholar]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev 2011, 111, 5922–5943. [Google Scholar]
- Odusanwo, O.; Chinthamani, S.; McCall, A.; Duffey, M.E.; Baker, O.J. Resolvin D1 prevents TNF-α-mediated disruption of salivary epithelial formation. Am. J. Physiol. Cell. Physiol 2012, 302, C1331–C145. [Google Scholar]
- Eickmeier, O.; Seki, H.; Haworth, O.; Hilberath, J.N.; Gao, F.; Uddin, M.; Croze, R.H.; Carlo, T.; Pfeffer, M.A.; Levy, B.D. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal. Immunol. 2012. [Google Scholar] [CrossRef]
- Clària, J.; Dalli, J.; Yacoubian, S.; Gao, F.; Serhan, C.N. Resolvin D1 and resolvin D2 govern local inflammatory tone in obese fat. J. Immunol. 2012. [Google Scholar] [CrossRef]
- Lee, C.H. Resolvins as new fascinating drug candidates for inflammatory diseases. Arch. Pharm. Res 2012, 35, 3–7. [Google Scholar]
- Uddin, M.; Levy, B.D. Resolvins: Natural agonists for resolution of pulmonary inflammation. Prog. Lipid Res 2011, 50, 75–88. [Google Scholar]
- Baek, S.H.; Seo, J.K.; Chae, C.B.; Suh, P.G.; Ryu, S.H. Identification of the peptides that stimulate the phosphoinositide, hydrolysis in lymphocyte cell lines from peptide libraries. J. Biol. Chem 1996, 271, 8170–8175. [Google Scholar]
- Dahlgren, C.; Christophe, T.; Boulay, F.; Madianos, P.N.; Rabiet, M.J.; Karlsson, A. The synthetic chemoattractant Trp-Lys-Tyr-Met-Val-DMet activates neutrophils preferentially through the lipoxin A(4) receptor. Blood 2000, 95, 1810–1818. [Google Scholar]
- Christophe, T.; Karlsson, A.; Dugave, C.; Rabiet, M.J.; Boulay, F.; Dahlgren, C. The Synthetic Peptide Trp-Lys-Tyr-Met-Val-Met-NH2 Specifically Activates Neutrophils through FPRL1/Lipoxin A4 Receptors and Is an Agonist for the Orphan Monocyte-expressed Chemoattractant Receptor FPRL2. J. Biol. Chem 2001, 276, 21585–21593. [Google Scholar]
- Le, Y.; Gong, W.; Li, B.; Dunlop, N.M.; Shen, W.; Su, S.B.; Ye, R.D.; Wang, J.M. Utilization of two seven-transmembrane, G protein-coupled receptors, formyl peptide receptor-like 1 and formyl receptor, by the synthetic hexapeptide WKYMVm for human phagocyte activation. J. Immunol 1999, 163, 6777–6784. [Google Scholar]
- Kang, H.K.; Lee, H.Y.; Kim, M.K.; Park, K.S.; Park, Y.M.; Kwak, J.Y.; Bae, Y.S. The synthetic peptide Trp-Lys-Tyr-Met-Val-D-Met inhibits human monocyte-derived dendritic cell maturation via formyl peptide receptor and formyl peptide receptorlike 2. J. Immunol 2005, 175, 685–692. [Google Scholar]
- Bae, Y.S.; Kim, Y.; Kim, Y.; Kim, J.H.; Suh, P.G.; Ryu, S.H. Trp-Lys-Tyr-Met-Val-D-Met is a chemoattractant for human phagocytic cells. J. Leukoc. Biol 1999, 66, 915–922. [Google Scholar]
- Karlsson, J.; Fu, H.M.; Boulay, F.; Bylund, J.; Dahlgren, C. The peptide Trp-Lys-Tyr-Met-Val-D-Met activates neutrophils through the formyl peptide receptor only when signaling through the formylpeptide receptor like 1 is blocked. A receptor switch with implications for signal transduction studies with inhibitors and receptor antagonists. Biochem. Pharmacol 2006, 71, 1488–1496. [Google Scholar]
- Lee, H.Y.; Jo, S.H.; Lee, C.; Baek, S.H.; Bae, Y.S. Differential production of leukotriene B4 or prostaglandin E2 by WKYMVm or serum amyloid A via formyl peptide receptor-like 1. Biochem. Pharmacol 2006, 72, 860–868. [Google Scholar]
- Bae, Y.S.; Ju, S.A.; Kim, J.Y.; Seo, J.K.; Baek, S.H.; Kwak, J.Y.; Kim, B.S.; Suh, P.G. Trp-Lys-Tyr-Met-Val-D-Met stimulates superoxide generation and killing of Staphylococcus aureus via phospholipase D activation in human monocytes. J. Leukoc. Biol 1999, 65, 241–248. [Google Scholar]
- Shin, M.H.; Lee, Y.A.; Bae, Y.S.; Kita, H.; Kim, Y.; Ryu, S.H. The Synthetic Chemoattractant Peptide WKYMVm Induces Superoxide Production by Human Eosinophils via the Phosphoinositide 3-Kinase-Mediated Activation of ERK1/2. Int. Arch. Allergy Immunol 2005, 137, 21–26. [Google Scholar]
- Baek, S.H.; Bae, Y.S.; Seo, J.K.; Lee, Y.H.; Kim, J.H.; Kwun, K.B.; Suh, P.G.; Ryu, S.H. Trp-Lys-Tyr-Met-Val-Met activates mitogen-activated protein kinase via a PI-3 kinase-mediated pathway independent of PKC. Life Sci 1999, 65, 1845–1856. [Google Scholar]
- Lee, H.Y.; Kang, H.K.; Yoon, H.R.; Kwak, J.Y.; Bae, Y.S. Lysophosphatidic acid is a mediator of Trp-Lys-Tyr-Met-Val-D-Met-induced calcium influx. Biochem. Biophys. Res. Commun 2004, 324, 458–465. [Google Scholar]
- Kim, S.D.; Kim, J.M.; Jo, S.H.; Lee, H.Y.; Lee, S.Y.; Shim, J.W.; Seo, S.K.; Yun, J.; Bae, Y.S. Functional Expression of Formyl Peptide Receptor Family in Human NK Cells. J. Immunol 2009, 183, 5511–5517. [Google Scholar]
- Ammendola, R.; Russo, L.; De Felice, C.; Esposito, F.; Russo, T.; Cimino, F. Low-affinity receptor-mediated induction of superoxide by N-Formyl-Methionyl-Leucyl-Phenyalanine and WKYMVm in IMR90 human fibroblasts. Free Radic. Biol. Med 2004, 36, 189–200. [Google Scholar]
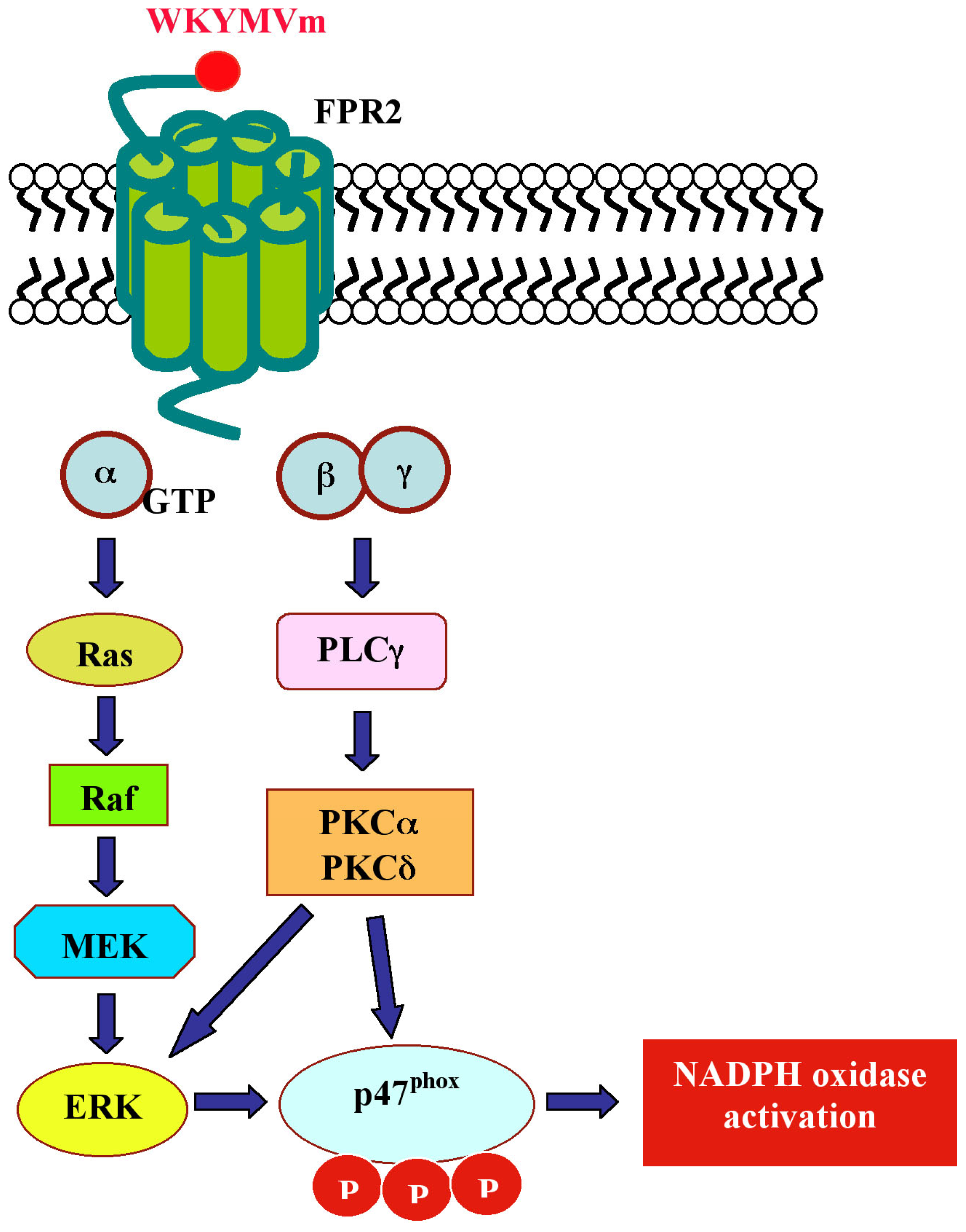
- Iaccio, A.; Collinet, C.; Gesualdi, N.M.; Ammendola, R. Protein kinase C-α and -δ are required for NADPH oxidase activation in WKYMVm-stimulated IMR90 human fibroblasts. Arch. Biochem. Biophys 2007, 459, 288–294. [Google Scholar]
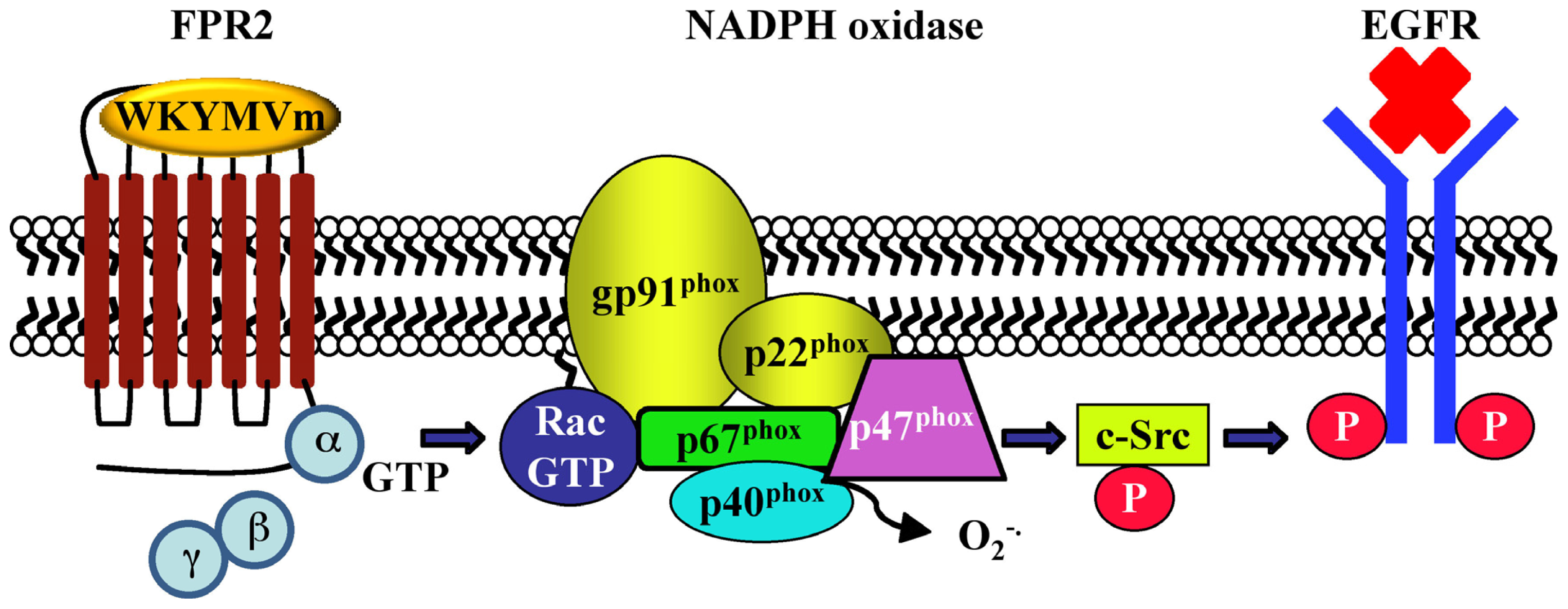
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med 2011, 51, 1126–1136. [Google Scholar]
- Kam, A.Y.; Tse, T.T.; Kwan, D.H.; Wong, Y.H. Formyl peptide receptor like 1 differentially requires mitogen-activated protein kinases for the induction of glial fibrillary acidic protein and interleukin-1α in human U87 astrocytoma cells. Cell. Signal 2007, 19, 2106–2117. [Google Scholar]
- Kam, A.Y.; Liu, A.M.; Wong, Y.H. Formyl peptide-receptor like-1 requires lipid raft and extracellular signal-regulated protein kinase to activate inhibitor-κB kinase in human U87 astrocytoma cells. J. Neurochem 2007, 103, 1553–1566. [Google Scholar]
- Kwan, D.H.; Kam, A.Y.; Wong, Y.H. Activation of the Human FPRL-1 Receptor Promotes Ca2+ Mobilization in U87 Astrocytoma Cells. Neurochem. Res 2008, 33, 125–133. [Google Scholar]
- Le, Y.; Wetzel, M.A.; Shen, W.; Gong, W.; Rogers, T.J.; Henderson, E.E.; Wang, J.M. Desensitization of Chemokine Receptor CCR5 in Dendritic Cells at the Early Stage of Differentiation by Activation of Formyl Peptide Receptors. Clin. Immunol 2001, 99, 365–372. [Google Scholar]
- Li, B.Q.; Wetzel, M.A.; Mikovits, J.A.; Henderson, E.E.; Rogers, T.J.; Gong, W.; Le, Y.; Ruscetti, F.W.; Wang, J.M. The synthetic peptide WKYMVm attenuates the function of the chemokine receptors CCR5 and CXCR4 through activation of formyl peptide receptor-like 1. Blood 2001, 97, 2941–2947. [Google Scholar]
- Kim, S.D.; Kim, Y.K.; Lee, H.Y.; Kim, Y.S.; Jeon, S.G.; Baek, S.H.; Song, D.K.; Ryu, S.H.; Bae, Y.S. The Agonists of Formyl Peptide Receptors Prevent Development of Severe Sepsis after Microbial Infection. J. Immunol 2010, 185, 4302–4310. [Google Scholar]
- Jo, E.J.; Lee, H.Y.; Kim, J.I.; Kang, H.K.; Lee, Y.N.; Kwak, J.Y.; Bae, Y.S. Activation of formyl peptide receptor-like 1 by WKYMVm induces serine phosphorylation of STAT3, which inhibits its tyrosine phosphorylation and nuclear translocation induced by hydrogen peroxide. Life Sci 2004, 75, 221–2232. [Google Scholar]
- Klein, C.; Paul, J.I.; Sauve, K.; Schmidt, M.M.; Arcangeli, L.; Ransom, J.; Trueheart, J.; Manfredi, J.P.; Broach, J.R.; Murphy, A.J. Identification of surrogate agonists for the human FPRL-1 receptor by autocrine selection in yeast. Nat. Biotechnol 1998, 16, 1334–1337. [Google Scholar]
- Karlsson, J.; Stenfeldt, A.L.; Rabiet, M.J.; Bylund, J.; Forsman, H.F.; Dahlgren, C. The FPR2-specific ligand MMK-1 activates the neutrophil NADPH-oxidase, but triggers no unique pathway for opening of plasma membrane calcium channels. Cell Calcium 2009, 45, 431–438. [Google Scholar]
- Prat, C.; Haas, P.J.; Bestebroer, J.; de Haas, C.J.; van Strijp, J.A.; van Kessel, K.P. A homolog of formyl peptide receptor-like 1 (FPRL1) inhibitor from Staphylococcus aureus (FPRL1 inhibitory protein) that inhibits FPRL1 and FPR. J. Immunol 2009, 183, 6569–6578. [Google Scholar]
- Prat, C.; Bestebroer, J.; de Haas, C.J.; van Strijp, J.A.; van Kessel, K.P. A new staphylococcal anti-inflammatory protein that antagonizes the formyl peptide receptor-like 1. J. Immunol 2006, 177, 8017–8026. [Google Scholar]
- Hecht, I.; Rong, J.; Sampaio, A.L.; Hermesh, C.; Rutledge, C.; Shemesh, R.; Toporik, A.; Beiman, M.; Dassa, L.; Niv, H.; et al. A Novel Peptide Agonist of Formyl-Peptide Receptor-Like 1 (ALX) Displays Anti-Inflammatory and Cardioprotective Effects. J. Pharmacol. Exp. Ther 2009, 328, 426–434. [Google Scholar]
- Bae, G.H.; Lee, H.Y.; Jung, Y.S.; Shim, J.W.; Kim, S.D.; Baek, S.H.; Kwon, J.Y.; Park, J.S.; Bae, Y.S. Identification of novel peptides that stimulate human neutrophils. Exp. Mol. Med 2012, 44, 130–137. [Google Scholar]
- Nanamori, M.; Cheng, X.; Mei, J.; Sang, H.; Xuan, Y.; Zhou, C.; Wang, M.W.; Ye, R.D. A novel nonpeptide ligand for formyl peptide receptor-like 1. Mol. Pharmacol 2004, 66, 1213–1222. [Google Scholar]
- He, M.; Cheng, N.; Gao, W.W.; Zhang, M.; Zhang, Y.Y.; Ye, R.D.; Wang, M.W. Characterization of Quin-C1 for its anti-inflammatory property in a mouse model of bleomycin-induced lung injury. Acta Pharmacol. Sin 2011, 32, 601–610. [Google Scholar]
- Burli, R.W.; Xu, H.; Zou, X.; Muller, K.; Golden, J.; Frohn, M.; Adlam, M.; Plant, M.H.; Wong, M.; McElvain, M.; et al. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg. Med. Chem. Lett 2006, 16, 3713–3718. [Google Scholar]
- Schepetkin, I.A.; Kirpotina, L.N.; Tian, J.; Khlebnikov, A.I.; Ye, R.D.; Quinn, M.T. Identification of Novel Formyl Peptide Receptor-Like 1 Agonists That Induce Macrophage Tumor Necrosis Factor α Production. Mol. Pharmacol 2008, 74, 392–402. [Google Scholar]
- Cilibrizzi, A.; Quinn, M.T.; Kirpotina, L.N.; Schepetkin, I.A.; Holderness, J.; Ye, R.D.; Rabiet, M.J.; Biancalani, C.; Cesari, N.; Graziano, A.; et al. 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones: A novel class of small-molecule agonists for formyl peptide receptors. J. Med. Chem 2009, 52, 5044–5057. [Google Scholar]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Jutila, M.A.; Quinn, M.T. Gastrin-Releasing Peptide/Neuromedin B Receptor Antagonists PD176252, PD168368, and Related Analogs Are Potent Agonists of Human Formyl-Peptide Receptors. Mol. Pharmacol 2011, 79, 77–90. [Google Scholar]
- Ray, R.; Zhang, Z.; Lee, Y.C.; Gao, J.L.; Mukherjee, A.B. Uteroglobin suppresses allergen-induced TH2 differentiation by down-regulating the expression of serum amyloid A and SOCS-3 genes. FEBS Lett 2006, 580, 6022–6026. [Google Scholar]
- Svensson, L.; Redvall, E.; Bjorn, C.; Karlsson, J.; Bergin, A.M.; Rabiet, M.J.; Dahlgren, C.; Wenneras, C. House dust mite allergen activates human eosinophils via formyl peptide receptor and formyl peptide receptor-like 1. Eur. J. Immunol 2007, 37, 1966–1977. [Google Scholar]
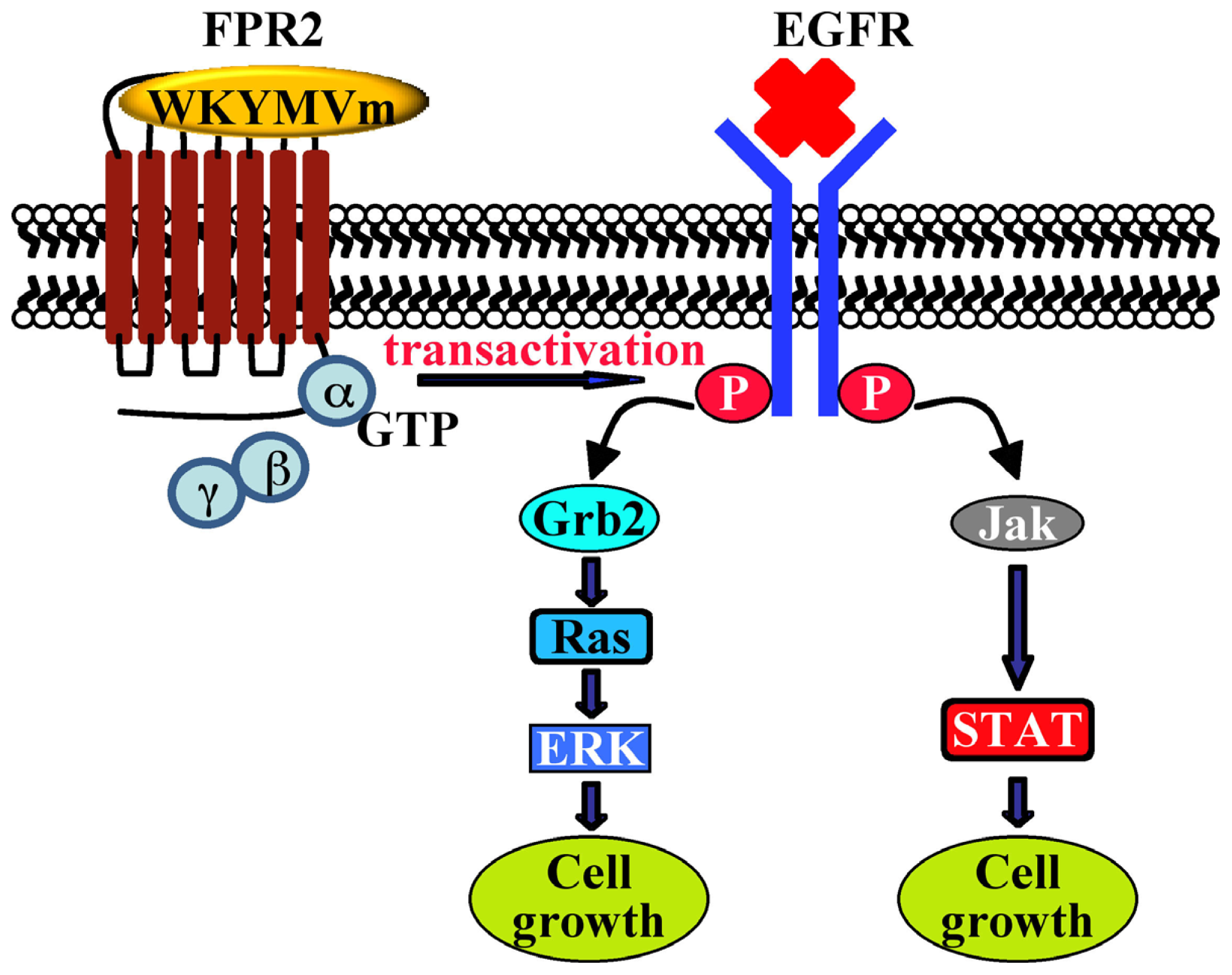

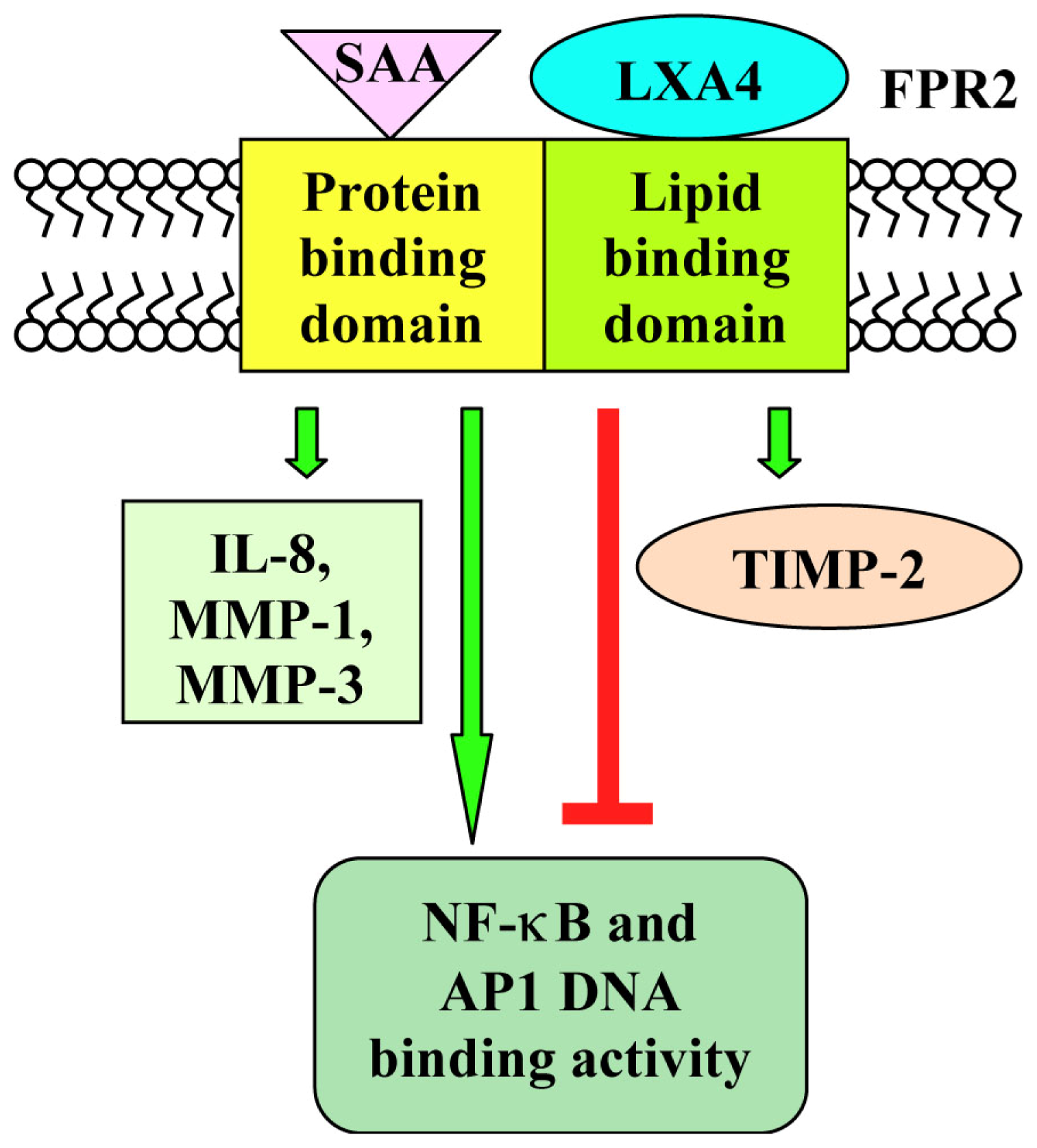{kind=link}
{kind=link}
{kind=link}
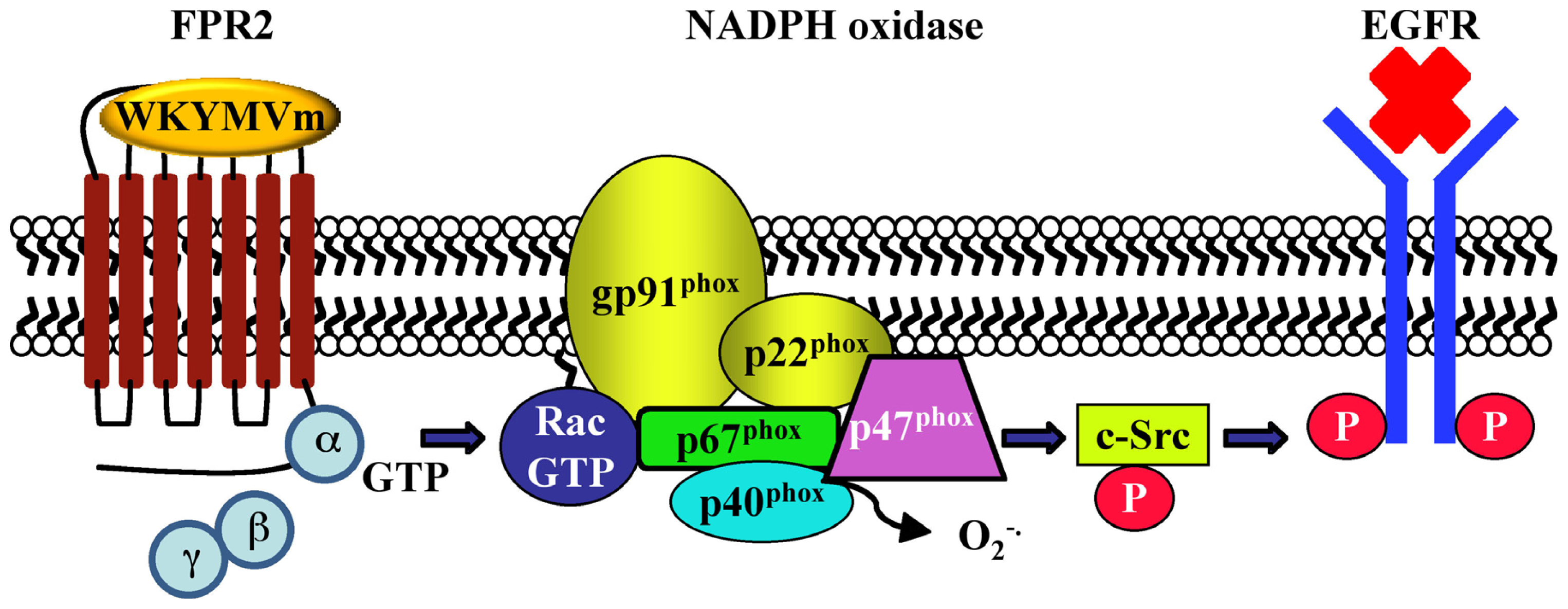{kind=link}
{kind=link}
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| Hp(2–20) | H. pylori | FPR2, FPR3 FPR2 | Mon.; Lymph. MKN-28, AGS | O2−. generation; apoptosis; chemotaxis; proliferation; VEGF secretion; ERKs, Akt and STAT3 activation | [6,9] | pEC50 = 6.52 |
| F peptide | HIV-1 | FPR2 | Mon.; Neutr. | chemotaxis; Ca2+ mobilization; desensitization CCR5 and CXCR4 | [7,10] | pEC50 = 5.00 |
| V3 peptide | HIV-1 | FPR2 | Mon.; Neutr. | chemotaxis; Ca2+ mobilization; desensitization CCR5 | [11] | pEC50 = 5.82 |
| T21/DP107 | HIV-1 | FPR2 | Mon.; Neutr. | chemotaxis; Ca2+ mobilization; | [12] | pEC50 = 6.30 |
| N36 peptide | HIV-1 | FPR2 | Mon.; Neutr. | chemotaxis; Ca2+ mobilization; desensitization chemokine receptors; increased expression of TRAIL; NFκB activation; apoptosis | [13,14] | pEC50 = 5.00 |
| PSMs | Peptide toxins | FPR2 | Neutrophils | Ca++ mobilization; chemotaxis; IL-8 release; NADPH oxidase activation | [15,16] | pEC50 = 8.67 |
| Ligands | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| f-MLKLIV | Mitochondria | FPR1, FPR2 | FPR-transfected HL60 | chemotaxis; Ca2+ mobilization; ERKs activation | [8] | pEC50 = 7.92, 7.26 |
| f-MMYALF | Mitochondria | FPR2 > FPR1 | FPR-transfected HL60 | chemotaxis; O2−. generation | [8] | pEC50 = 7.82, 7.92 |
| MCT-2 | Mitochondria | FPR2 | Neutrophils; Granulocytes | chemotaxis; Ca2+ mobilization; ERKs activation β-hexosaminidase release | [18] | EC50 = 240 nM |
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| SAA | Acute-phase protein | FPR2 | Mon.; Neutr.; Lymph. | chemotaxis; Ca2+ mobilization; production of metalloproteases and cytokines; expression of cytokine receptors; ERKs, JNK and p38MAPK activation; COX2 and NF-κB induction; IL-8, IL-10 and TNF-α release; MMP-9 upregulation | [3,21] | pEC50 = 7.35 |
| FLS | proliferation; anti-apoptosis; ERKs and Akt activation; expression of MMP-1 and -3; IL-6 production | [25–27] | ||||
| Endothelial cells | production of PTX3 | [30] | ||||
| Aβ42 | Amyloid precursor | Mon.; RBL; FPR2/HEK293 | chemotaxis; Ca2+ mobilization; O2−. generation | [4,36] | pEC50 = 7.00 | |
| Glial cells | ERKs phosphorylation; PI3K/Akt pathway; PLD activation; FPR1/FPR2/MARCO physical and functional interaction | [35,37] | EC50 = 5 μM | |||
| HN | Neuroprotective peptide | Mononuclear phagocytes | Chemotaxis; Ca2+ mobilization; anti-apoptosis; ERKs phosphorylation | [38] | pEC50 = 8.46 | |
| PrP106–126 | Prion protein | Glial cells | protein tyrosine phosphorylation; IL-6 and TNF-α increase; chemotaxis; Ca2+ mobilization | [42] | pEC50 = 4.60 | |
| Ana-1 | Chemotaxis | [43] |
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| D2D388–274 | uPAR | FPR2 | THP-1; Mon. | chemotaxis; decreased chemokine-induced integrin-dependent cell adhesion | [5,48] | pEC50 = 7.08 |
| uPAR84–95 | uPAR | FPR2, FPR3 | Basophils | chemotaxis | [5] | Kd = 82.6 |
| LL-37 | Cathelicidin | FPR2 | FPR2/HEK293; Lymph.; Mon. | chemotaxis; Ca2+ mobilization | [51] | pEC50 = 6.00 |
| Neutrophils | Anti-apoptosis; Bcl-xL expression; inhibition of caspase 3 and of SAA-induced IL-8 production; inhibition of SAA-induced ERKs and p38MAPK activity; LTB4 production; cPLA2 phosphorylation | [52,53,59,60] | ||||
| Endothelial cells | vessel growth; Ca2+ mobilization; NF-κB nuclear translocation; PKC activation; O2−. generation; ERKs phosphorylation | [54] | ||||
| NCI-H292 | proliferation; migration; wound healing | [55] | ||||
| MSCs | inhibition of tumor growth; ERKs phosphorylation | [56] | ||||
| Ovarian cancer cells | MAPK and JAK/STAT signaling; expression of angiopoietin-like 3, C5, collagen type XVIII, EGF, FGF1, FPR2, LL-37, MMP-2, uPA | [57] | ||||
| Hepatocarcinoma | M-CSF and MCP-1 expression; ROS-MAPK-NFκB signaling | [58] | ||||
| IMR9 | O2−. generation; p47phox and ERKs phosphorylation | [61] | ||||
| sCKβ8-1 | Chemokine | FPR2 | PMN, FPR2/CHO-K1 | chemotaxis; Ca2+ mobilization | [62] | pEC50 = 9.00 |
| SHAAGtide | CCL23 | FPR2 | Mon.; Neutr. | chemotaxis; Ca2+ mobilization | [63] | pEC50 = 7.72 |
| VIP | Pleiotropic peptide | VPAC1, FPR2 | Monocytes | pro-inflammatory; PI3K/ERK activation; CD11b upregulation | [67] | |
| PACAP27 | Neuropeptide | FPR2 | Neutrophils | chemotaxis; Ca2+ mobilization; CD11b upregulation; ERKs, Akt, p38MAPK phosphorylation | [69] | EC50 = 0.33 μM |
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| antiflammin-2 | ANXA1 | FPR2 | FPR2/HEK293 | ERKs phosphorylation | [76] | EC50 = 1.2 μM |
| Ac2-26 | ANXA1 | FPR1 > FPR2 | PMN; FPR1/HE293; FPR2/HEK293 | ERKs phosphorylation | [73] | pEC50 = 6.05 EC50 = 25 μM |
| FPR2 | Synovial fibroblasts | MMP-1 secretion | [78] | |||
| FPR1 > FPR2 | MDA-MB-231 | Cell proliferation | [81] | |||
| ANXA1 | ANXA1 | FPR2 | PMN; FPR2/HEK293 | ERKs phosphorylation | [73] | EC50 = 0.15 μM EC50 = 25 μM |
| Synovial fibroblasts | MMP-1 secretion | [78] | ||||
| MCF-7 | PI3K/Akt/p70S6K pathway; cyclin D increase | [79] | ||||
| SKCO-15 | Cell invasion | [80] | ||||
| Lung fibroblasts | TNFα-induced cell proliferation; inflammatory responses; activation of ERK and NF-κB pathways | [82] |
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| TA | Antimicrobial peptide | FPR2 | Mon.; Neutr.; Macroph. | chemotaxis; Ca2+ mobilization; ERKs activation | [85] | pEC50 = 6.60 |
| I4S10-C | Antimicrobial peptide | FPR2 | FPR2/HEK293 | Cell migration | [85] | EC50 = 5 μM |
| I4G10-C | Antimicrobial peptide | FPR2 | FPR2/HEK293 | Cell migration | [85] | EC50 = 0.5 μM |
| Rana-6 | Antimicrobial peptide | FPR2 | FPR2/HEK293 | Cell migration | [85] | EC50 = 5 μM |
| L37pA | apoA-I | FPR2 | Mon.; FPR2/HEK293 | chemotaxis; Ca2+ mobilization; anti-inflammatory | [86] | EC50 = 112 nM |
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| LXA4 | Eicosanoids | FPR2 | Epith. and Endothel. cells; Neutrophils | NO production; inhibition of neutrophil infiltration and transmigration | [93–95] | pKd = 8.77 EC50 = 50 nM |
| Neutrophils | chemotaxis; Ca2+ mobilization; PKC-dependent PLD activation | [96] | ||||
| Renal mesangial cells | inhibition of LTD4- and LXA4-induced cell proliferation and PI3K activity; ERKs and p38MAPK phosphorylation; inhibition of PDGF-Rβ and EGF-R; p21cip1 and p27kip1 modulation; inhibition of PDGF-induced increase of CDK2/cyclin E complex; block of G1-S progression | [97,98] | ||||
| HLF | inhibition of CTGF-induced cell proliferation, of ERKs, PI3K and Akt phosphorylation, of cyclin D1 expression and of STAT3 DNA-binding activity; p27kip1 modulation | [99] | ||||
| MCF-7; MDA-MB-231 | increase in cyclin D1; Akt and p79S6K phosphorylation | [81] | ||||
| Synovial fibroblasts | inhibition of IL-1β-induced IL-6, IL-8 and MMP-3 synthesis of FPR2 expression; downregulation of IL-1β-induced AP1 and NF-κB DNA binding activity | [100,101] | ||||
| Dendritic cells | induction of SOCS-2 | [106] | ||||
| Rv | Lipid mediator | GPCR-32, FPR2 | Salivary cells | cell migration; polarity; inhibition of TNF-α-induced cytoskeletal disruption; modulation of PI3K/Akt pathway | [108] | - |
| D1 | FPR2 | Acute lung injury | decrement of IL-1β, IL-6, TNF-α and of NF-κB p65 translocation | [109] | ||
| FPR2 | Inflamed adipose tissue | secretion of adiponectin; decreased pro-inflammatory adipokine production | [110] |
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| WKYMVm | Peptide library | FPR2 | Neutrophils | increase in Ca++ concentration; NADPH oxidase activation; cPLA2-mediated arachidonic acid release; increase of LTB4 production | [115,120] | pEC50 = 8.70 |
| Monocytes | chemotaxis; p125FAK, Pyk, MEK, ERKs, Akt and RhoA phosphorylation; NADPH oxidase activation; PKC and PLD activation | [118,121] | EC50 = 50 nM | |||
| Eosinophils | ERKs phosphorylation; NADPH oxidase activation; PI3K/ERK pathway | [122] | ||||
| U937 | ERKs phosphorylation; G0/PI3K/Ras/Raf-1 pathway; cPLA and PLD activation; LPA formation; Ca++ influx | [123,124] | pEC50 = 10.13 | |||
| NK | chemotaxis in IL-2-activated NK cells; ERKs, p38MAPK and JNK activation | [125] | EC50 = 9.2 nM | |||
| IMR90 | ERKs activation; p47phox translocation; NADPH oxidase, PKCα and PKCδ activation | [126,127] | Kd = 155,99 nM | |||
| CaLu-6 | EGFR transactivation, p47phox phosphorylation, NADPH oxidase activation; c-Src activation; STAT3 pathway; cell growth | [128] | - | |||
| U87 | ERKs, p38MAPK and JNK activation; c-Src and PLCβ activation; GFAP and IL-1α upregulation; IKK phosphorylation; PI3K activation; Ca++ influx | [129–131] | EC50 = 50–100 nM | |||
| iDC | downregulation of CCR5; PKC activation | [132] | ||||
| Osteosarcoma | downregulation of CXCR4 | [133] | ||||
| Mouse model | Anti-apoptosis; enhanced production of IFN-γ, IL-12, IL-17 and TGF-β; reduced production of TNF-α, IL-1β and IL-6 | [134] | ||||
| FPR2/RBL-2H3 | ERKs phosphorylation; STAT3 serine phosphorylation; PLD activation | [135] |
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| MMK-1 | Peptide library | FPR2 | Neutrophils; Monocytes | Ca++ mobilization; chemotaxis; NADPH oxidase activation | [136,137] | pEC50 = 8.70 |
| CGEN-855A | Peptide library | FPR2, FPR3 | FPR2/3-expressing cells | Ca++ mobilization; increase of cell impedance index; anti-inflammatory | [140] | IC50 = 189 nM Ki = 54.1 nM |
| MMHWAM | Peptide library | FPR2 | Neutrophils; Monocytes | Ca++ mobilization; chemotaxis; PLC activation; NADPH oxidase activation | [141] | - |
| Ligand | Origin | Selectivity | Cells | Effects | Ref. | Potency |
|---|---|---|---|---|---|---|
| Quin-C1 | Combinatorial library | FPR2 | Neutrophils; FPR2-expressing cells | chemotaxis; β-glucuronidase secretion | [142] | pEC50 = 5.72 |
| FPR2/RBL | Ca2+ mobilization; ERKs activation | [142] | ||||
| Mouse model | anti-inflammatory; reduction of the expression of TNF-α, IL-1β, keratinocyte-derived chemokine, TGF-β1 and CXCL10 | [143] | ||||
| Pyrazolone 24/43 | Combinatorial library | FPR2 | FPR2-transfected cells | anti-inflammatory; Ca2+ mobilization | [144] | pIC50 = 7.36 |
| Aryl carboxylic acid hydrazide derivatives | Chemolibrary of drug-like molecules | FPR2 | Monocytes; Macrophages; Phagocytes | TNFα production; Ca2+ mobilization; reactive oxygen species production; chemotaxis | [145] | EC50 = 2 μM |
| Pyridazin-derivatives | Ligand-based drug design approach | FPR1/FPR2 | Neutrophils | Ca2+ mobilization; chemotaxis | [146] | EC50 = 13.1 μM |
| AG-26, AG-09/4-AG09/8 | Chemolibrary of drug-like molecules | FPR2 | FPR2-transfected RBL-2H3 | Ca2+ mobilization | [84] | EC50 = 0.5 μM, 0.3–12.6 μM |
| PD168368; PD176252; A-716223 | Screening of known GPCR ligands | FPR1/FPR2 | FPRs-transfected HL-60; Neutrophils | Ca2+ mobilization; reactive oxygen species production | [147] | EC50 = 0.5 μM, 0.9 μM, 18.3 μM |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct Signaling Cascades Elicited by Different Formyl Peptide Receptor 2 (FPR2) Agonists. Int. J. Mol. Sci. 2013, 14, 7193-7230. https://doi.org/10.3390/ijms14047193
Cattaneo F, Parisi M, Ammendola R. Distinct Signaling Cascades Elicited by Different Formyl Peptide Receptor 2 (FPR2) Agonists. International Journal of Molecular Sciences. 2013; 14(4):7193-7230. https://doi.org/10.3390/ijms14047193
Chicago/Turabian StyleCattaneo, Fabio, Melania Parisi, and Rosario Ammendola. 2013. "Distinct Signaling Cascades Elicited by Different Formyl Peptide Receptor 2 (FPR2) Agonists" International Journal of Molecular Sciences 14, no. 4: 7193-7230. https://doi.org/10.3390/ijms14047193