Analysis of Conformational Motions and Residue Fluctuations for Escherichia coli Ribose-Binding Protein Revealed with Elastic Network Models


Abstract
:1. Introduction
2. Results and Discussions
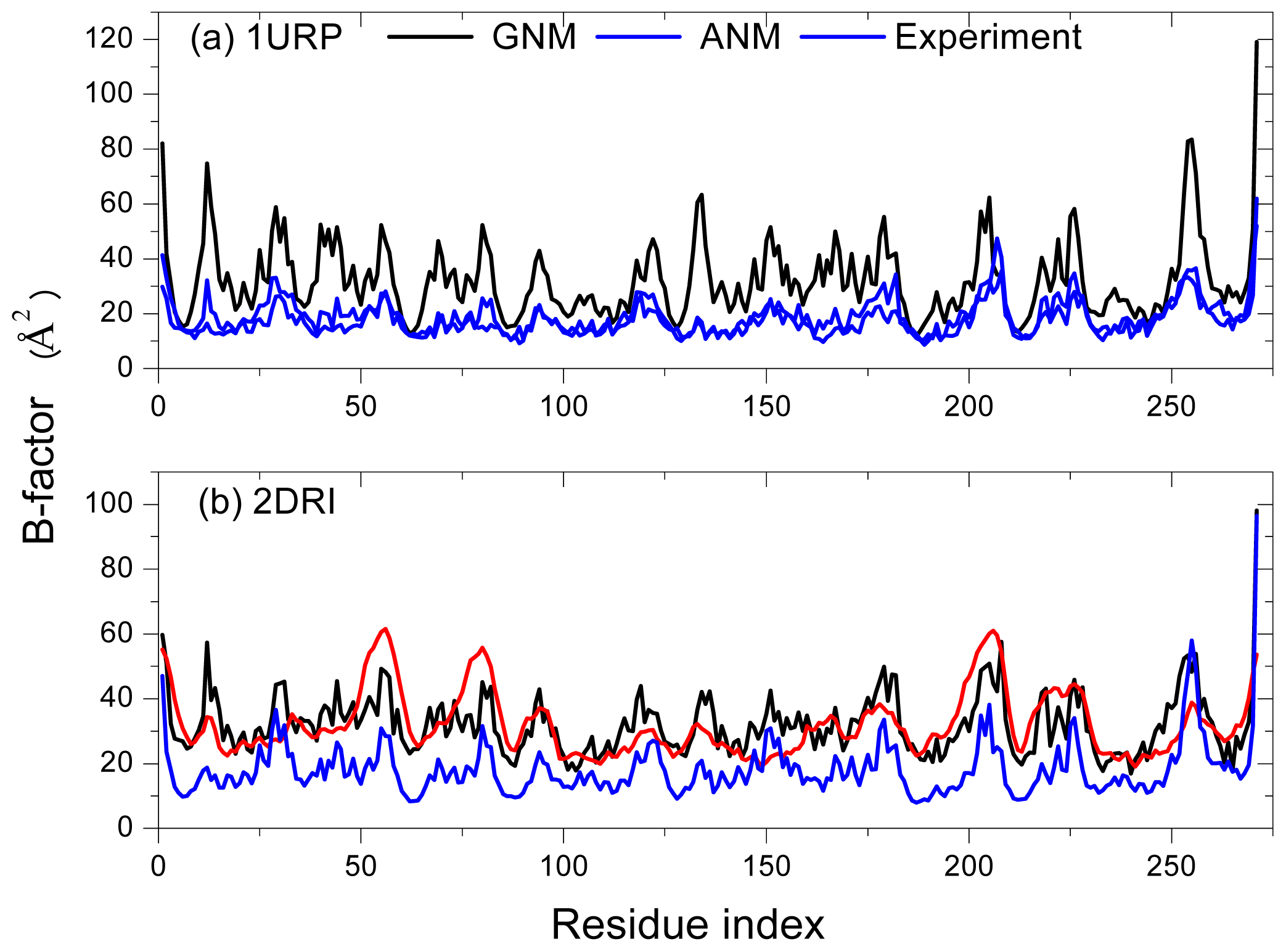
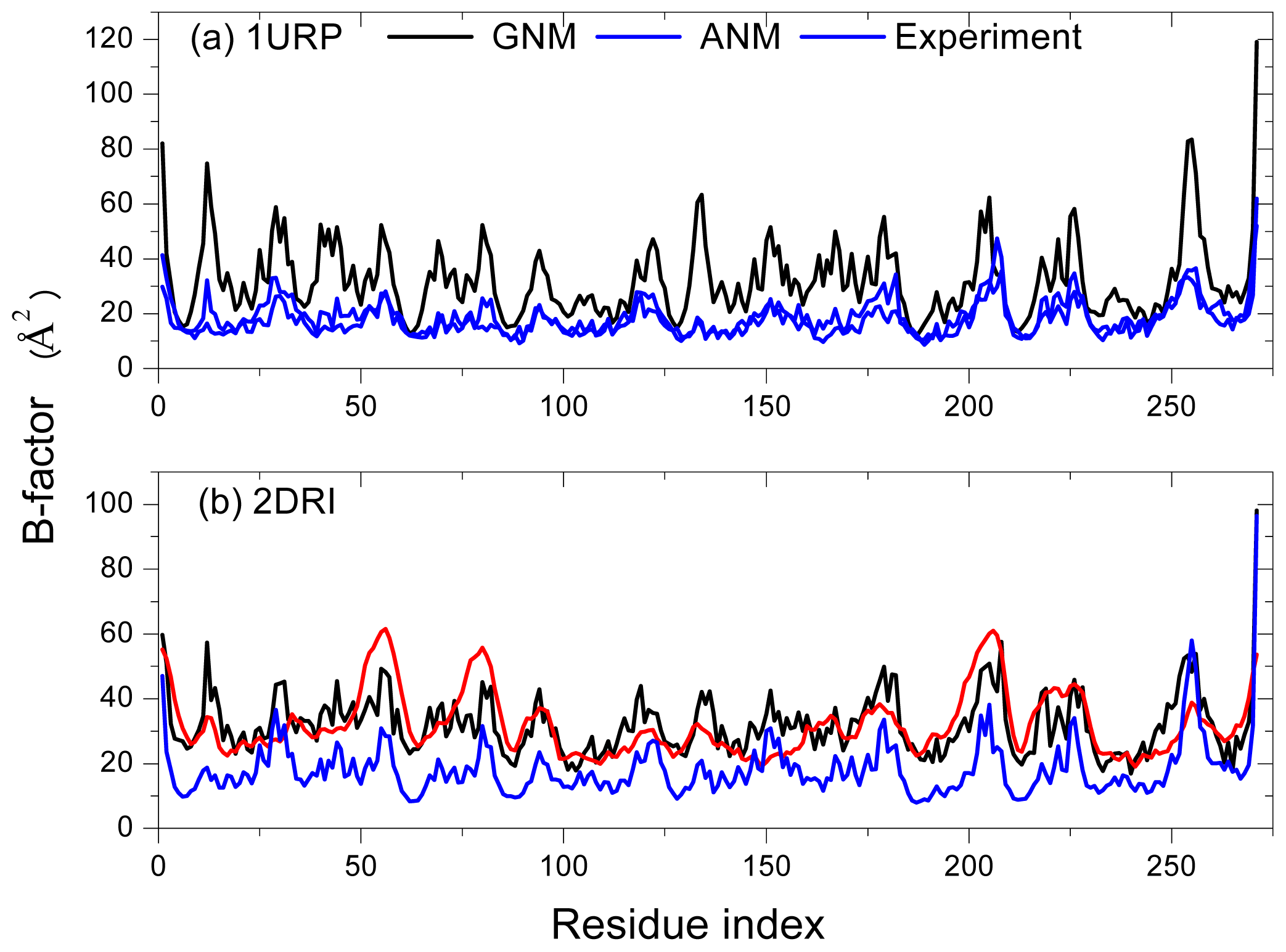
2.1. Comparison of Equilibrium Fluctuations of GNM and ANM with Experimental Temperature Factors
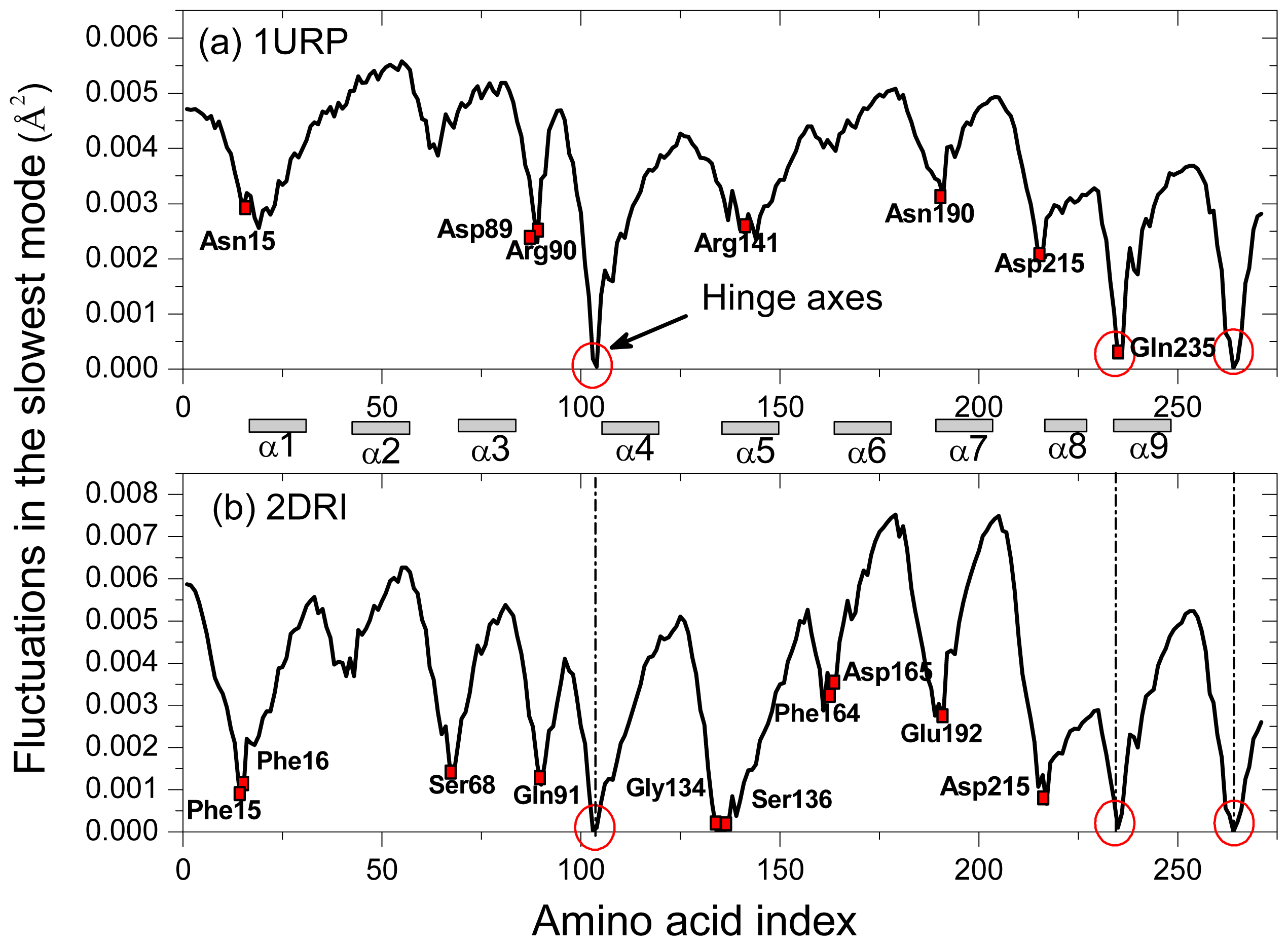
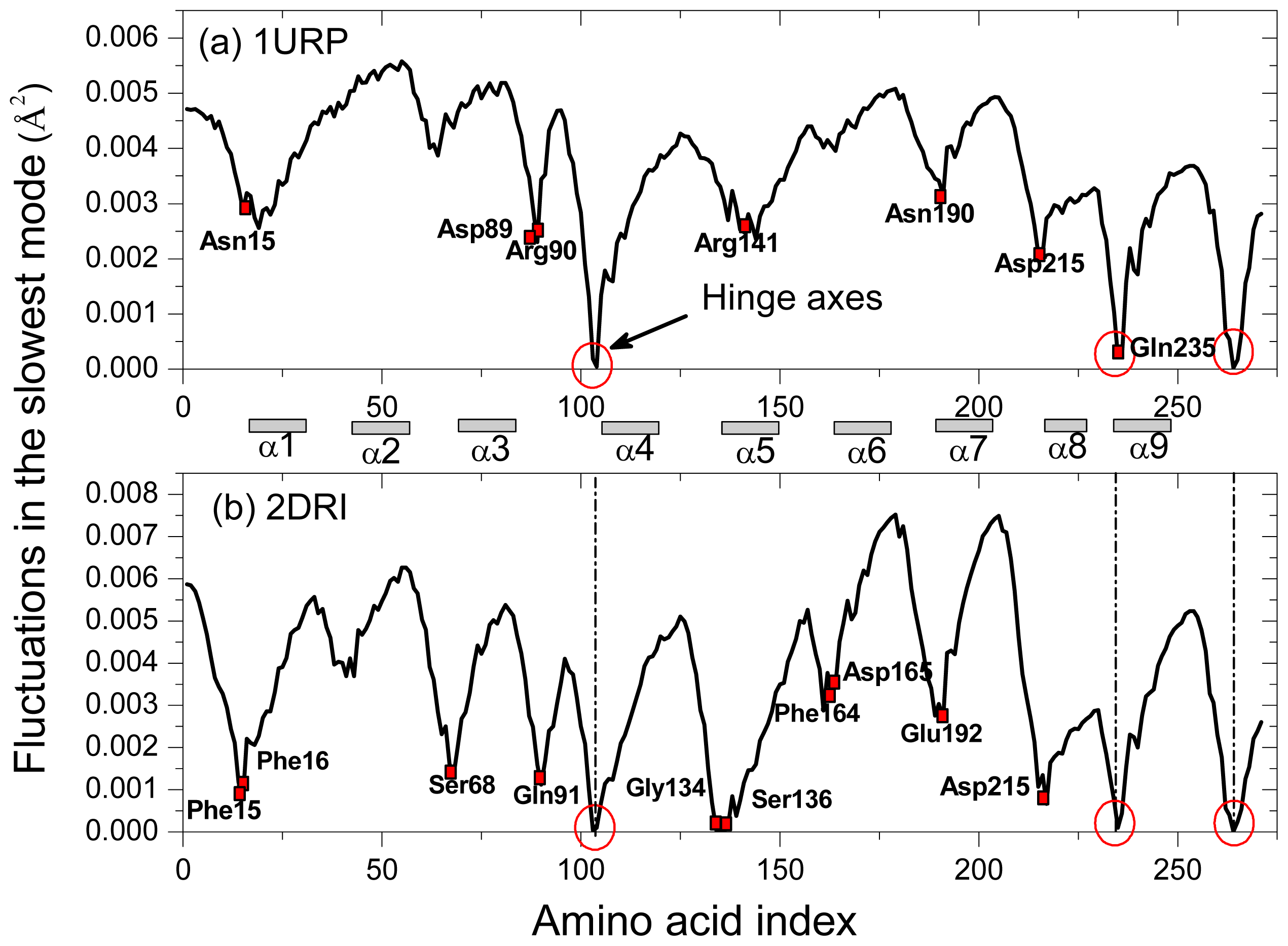
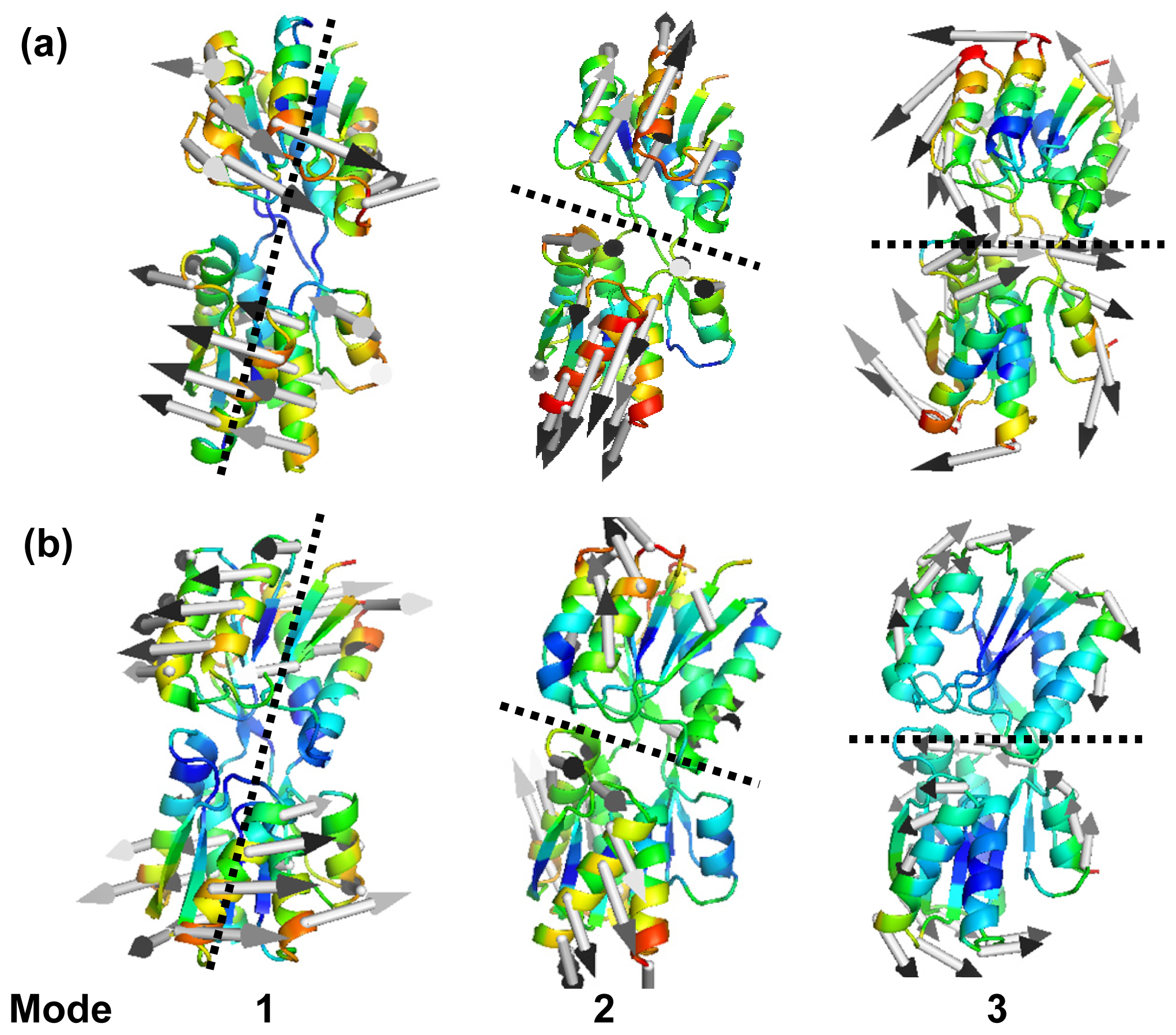
2.2. The Slow Mode of the Motion
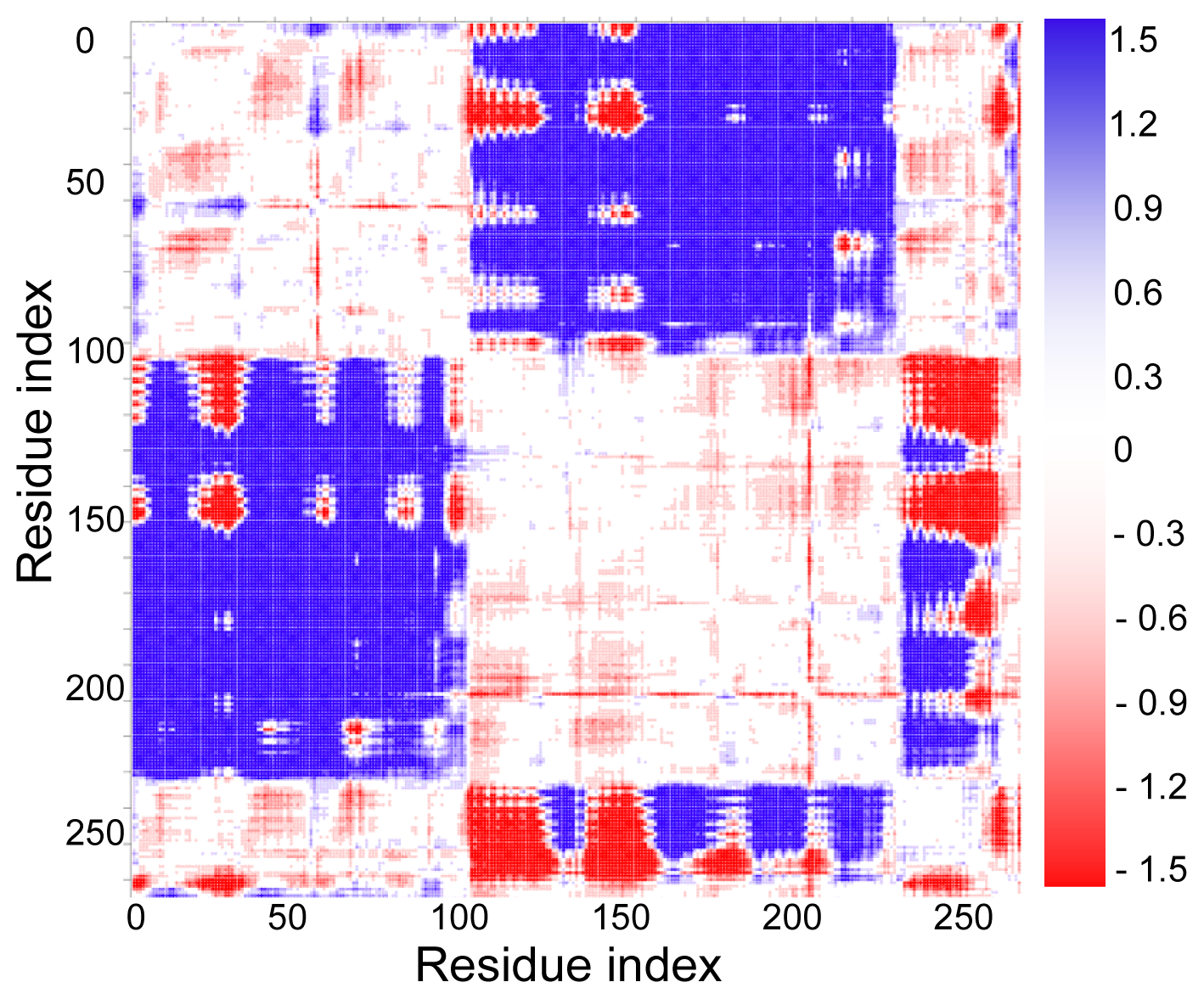
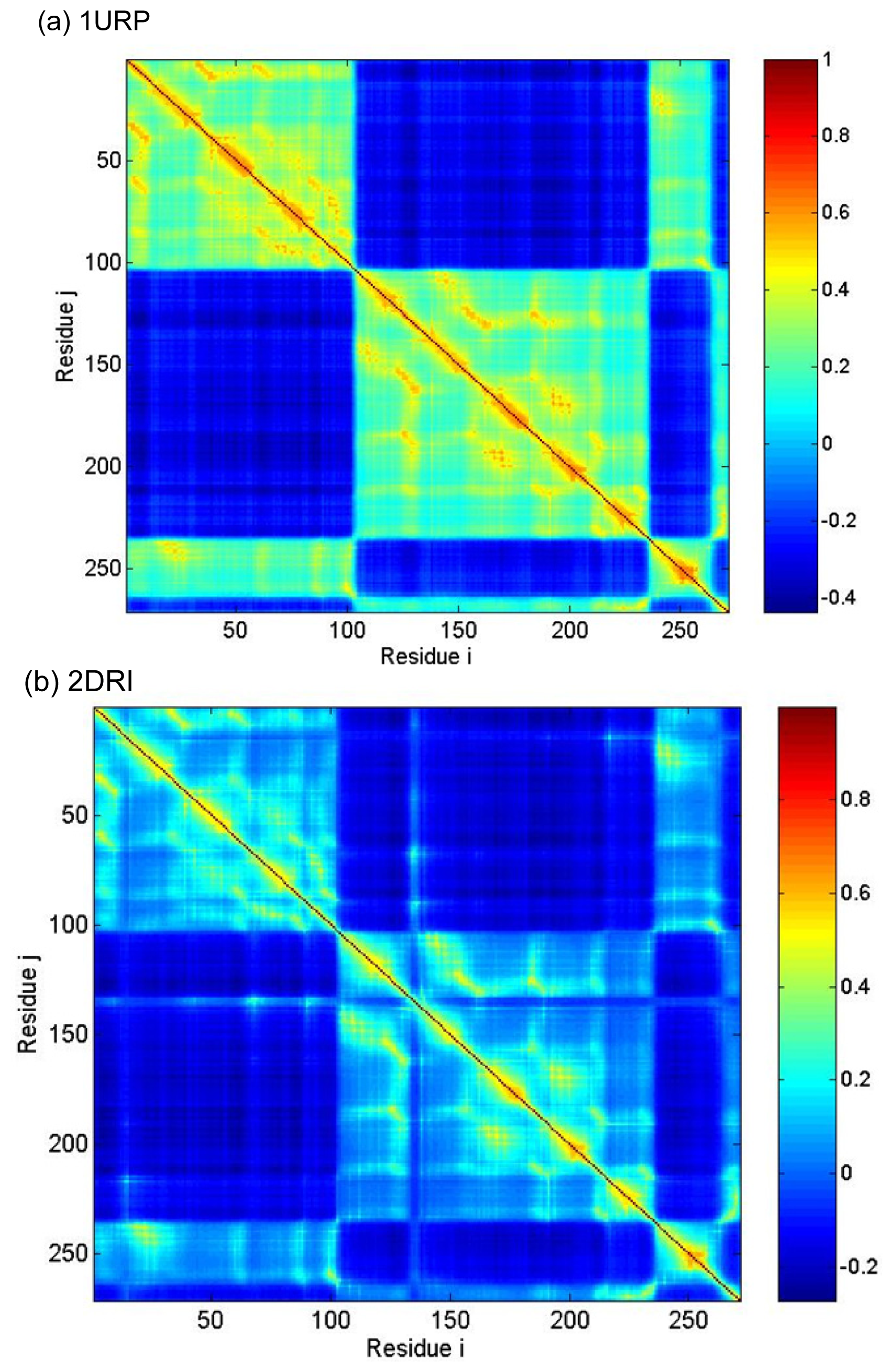
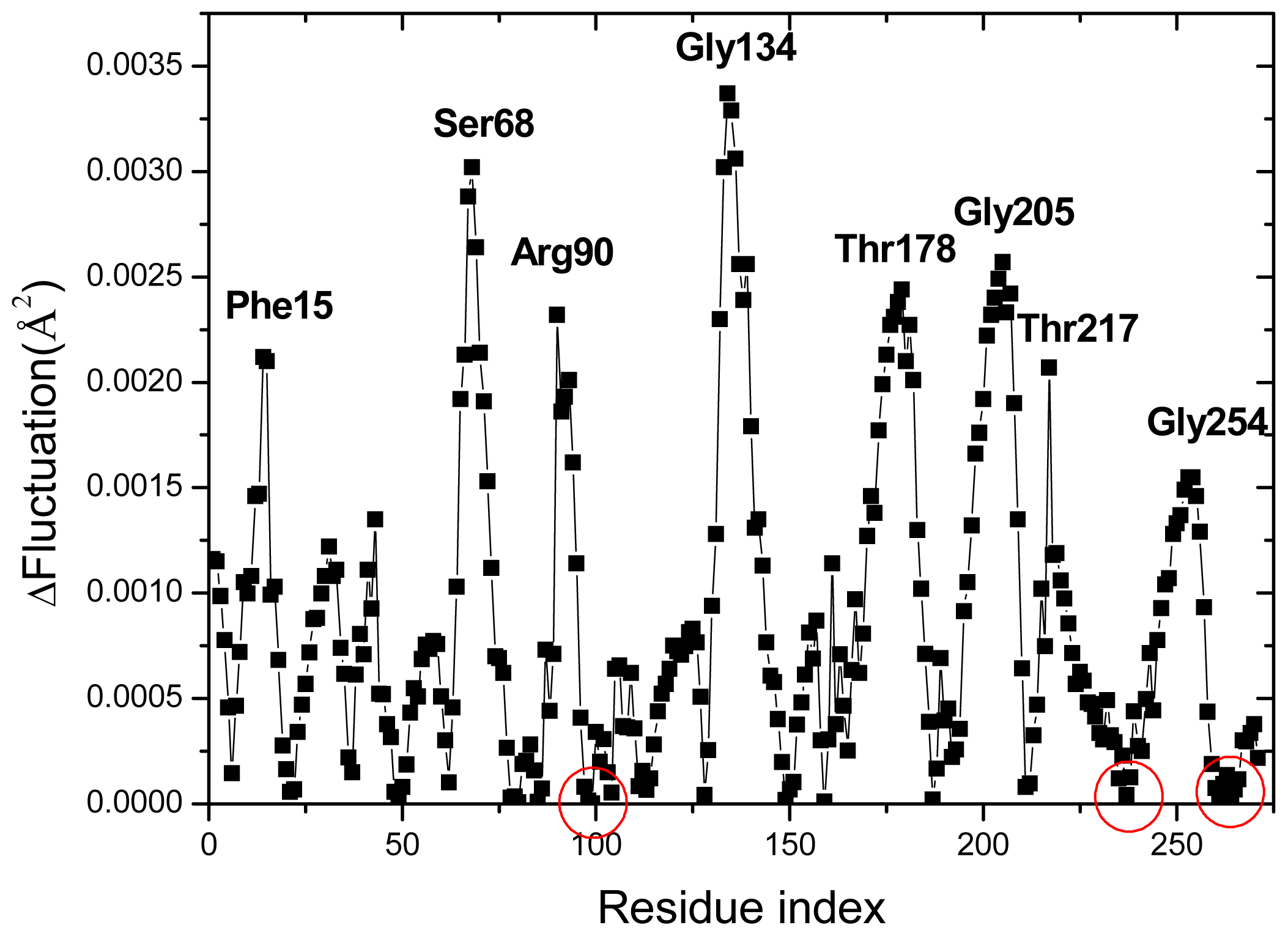
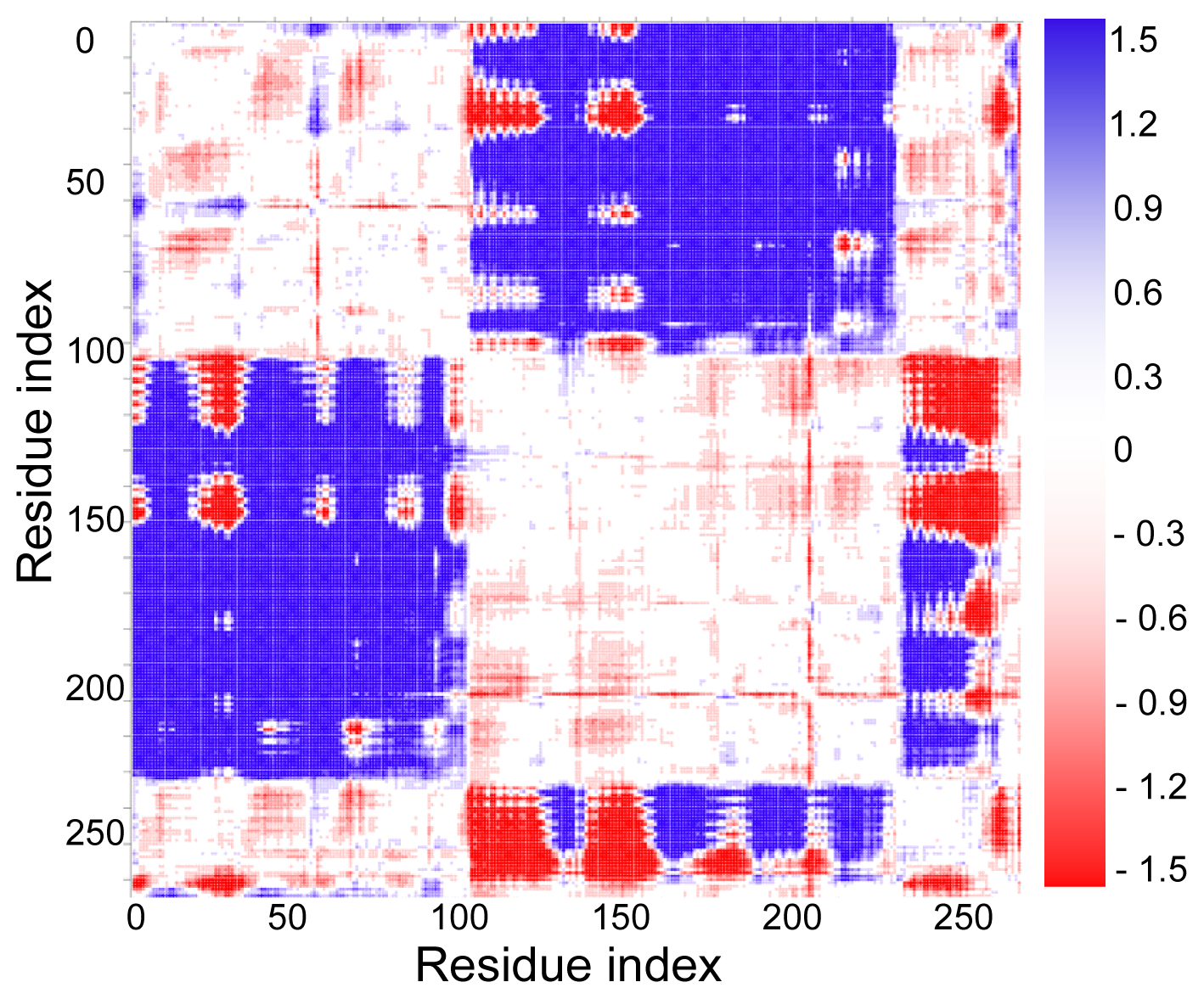
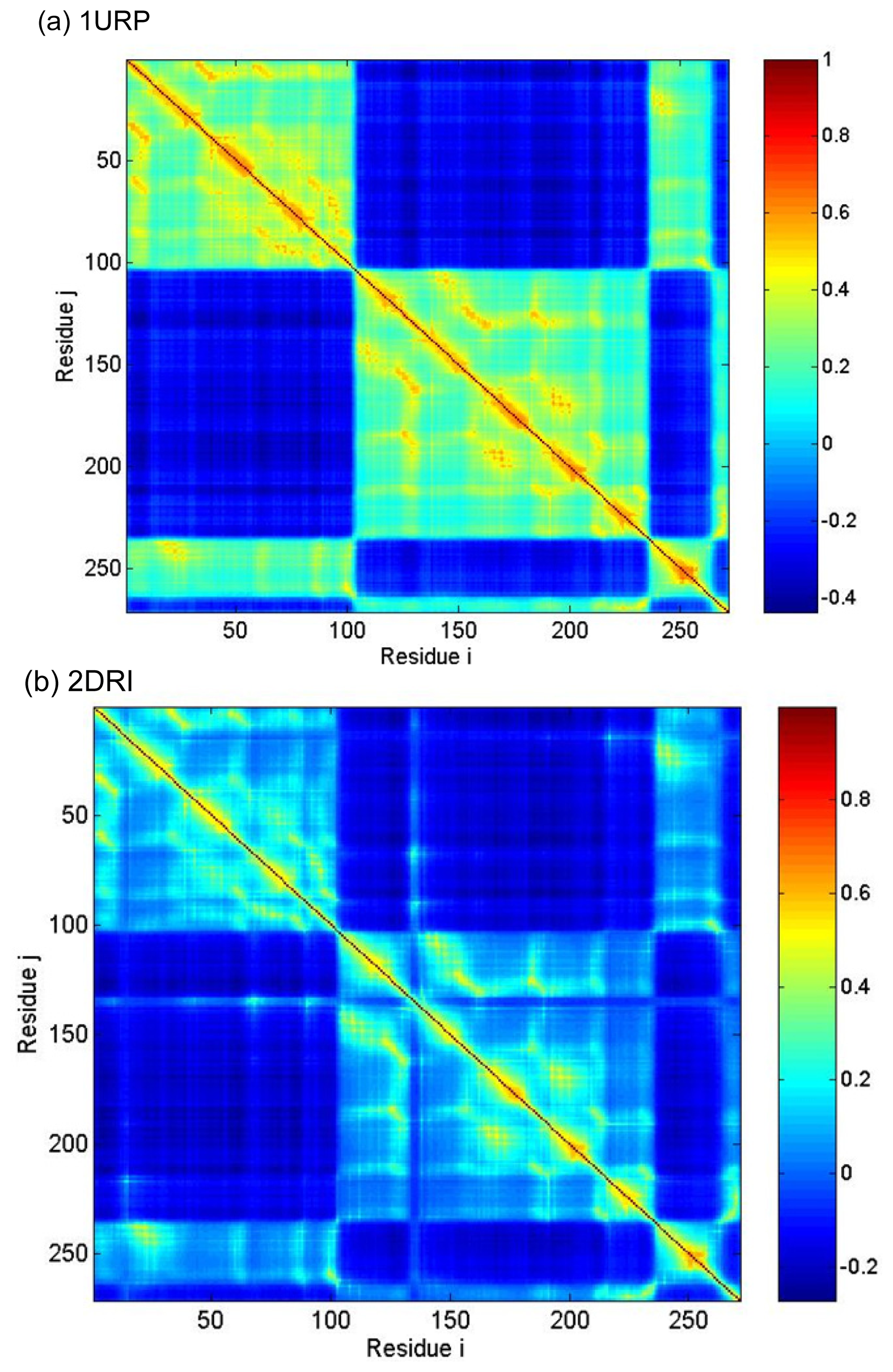
2.3. Cross-Correlations between Residue Fluctuations
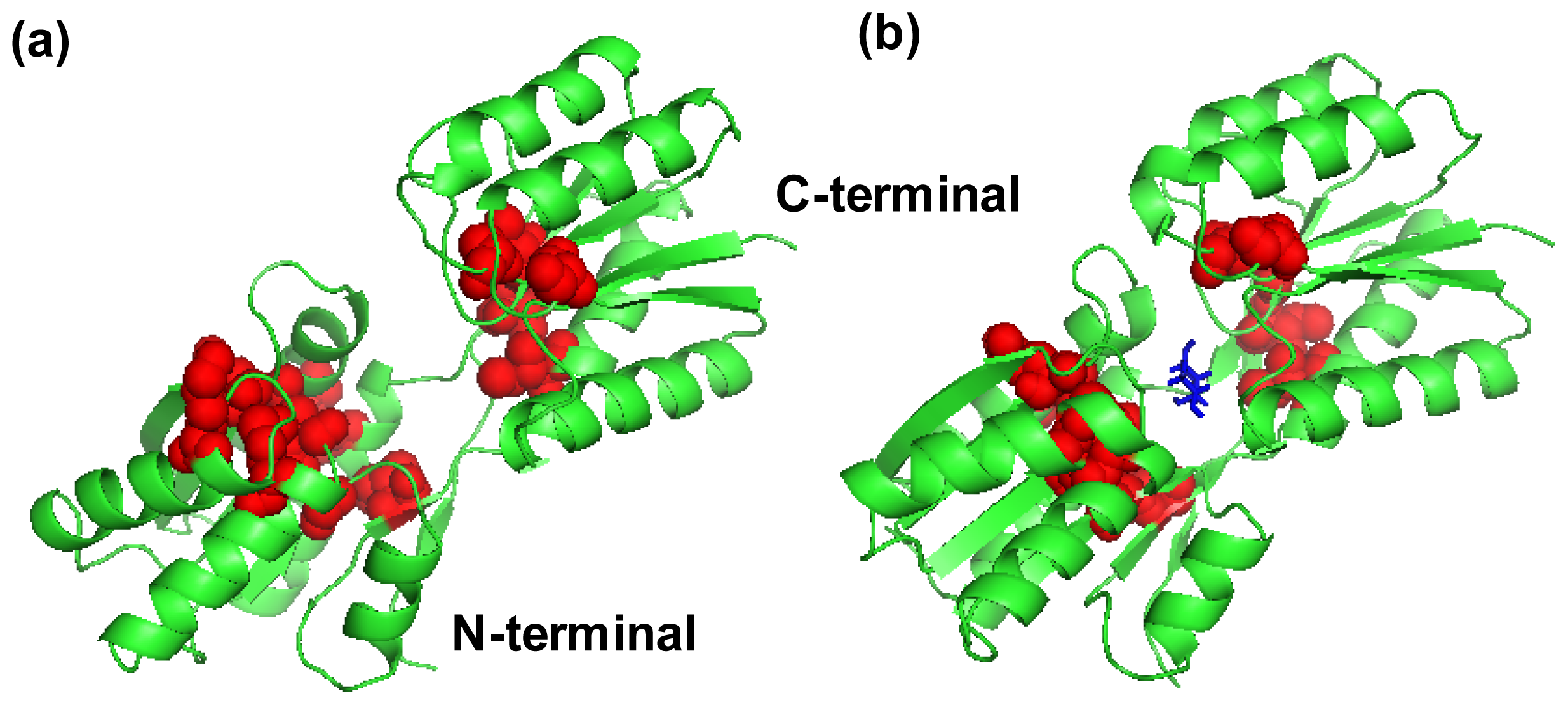
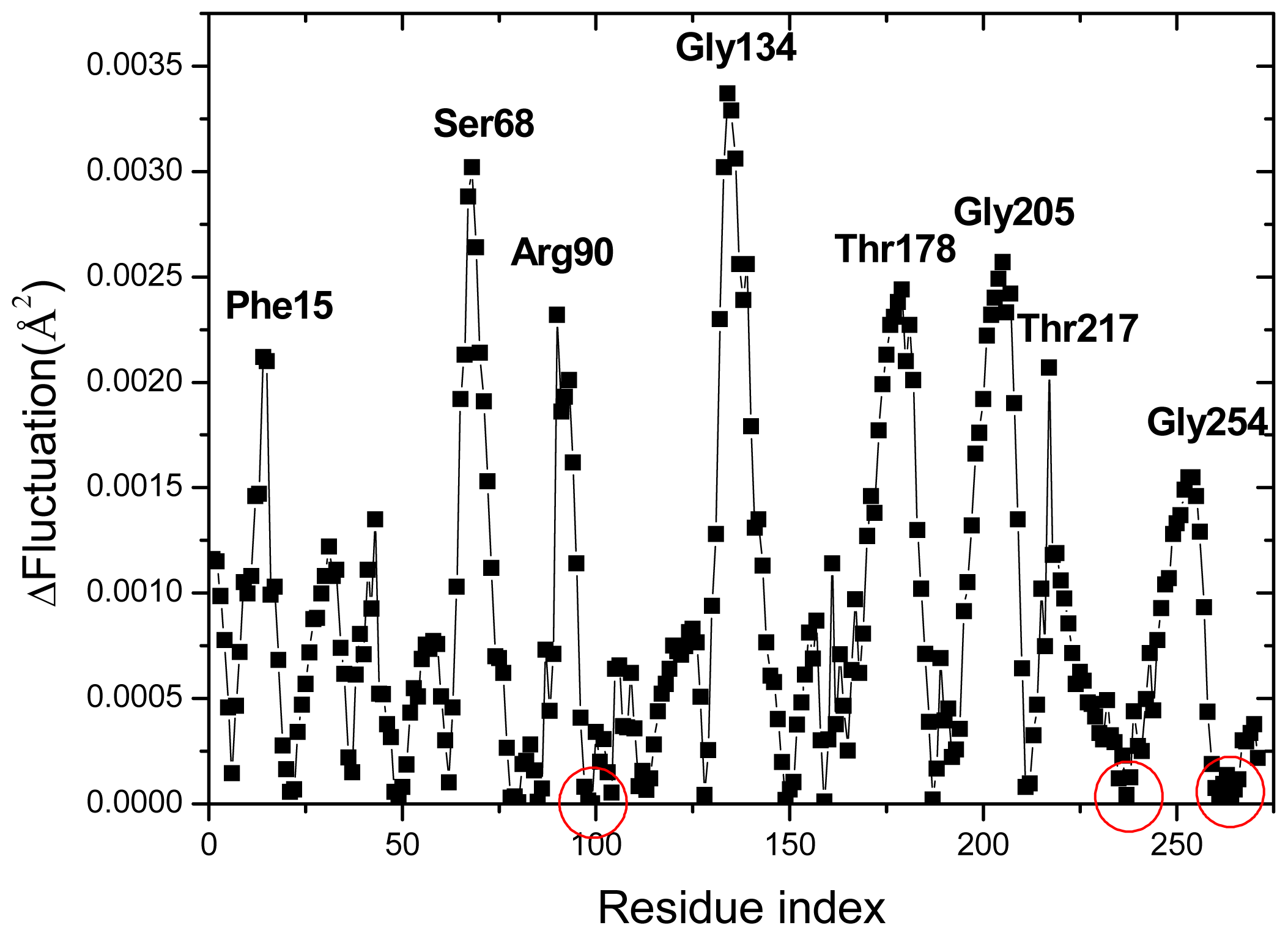
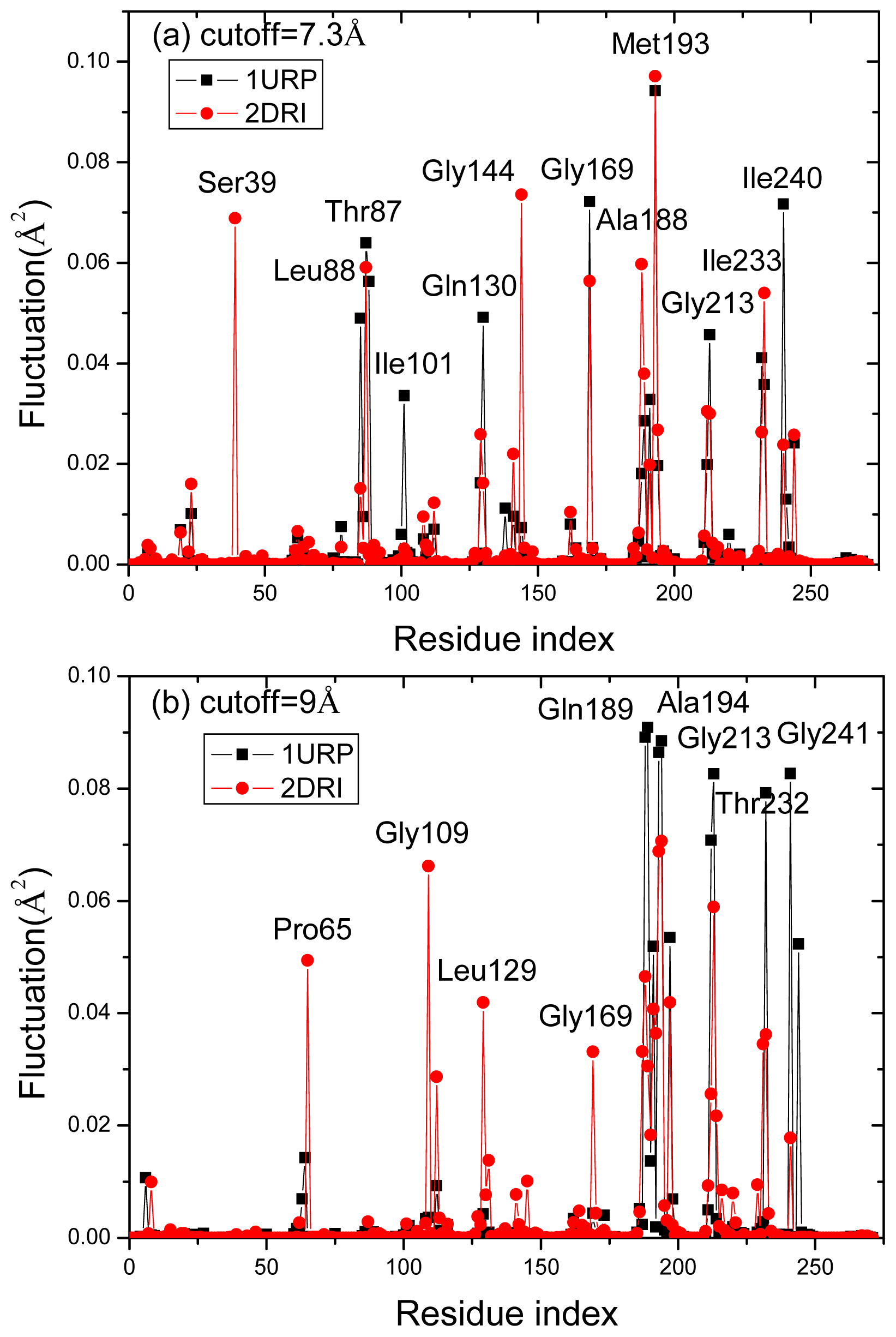
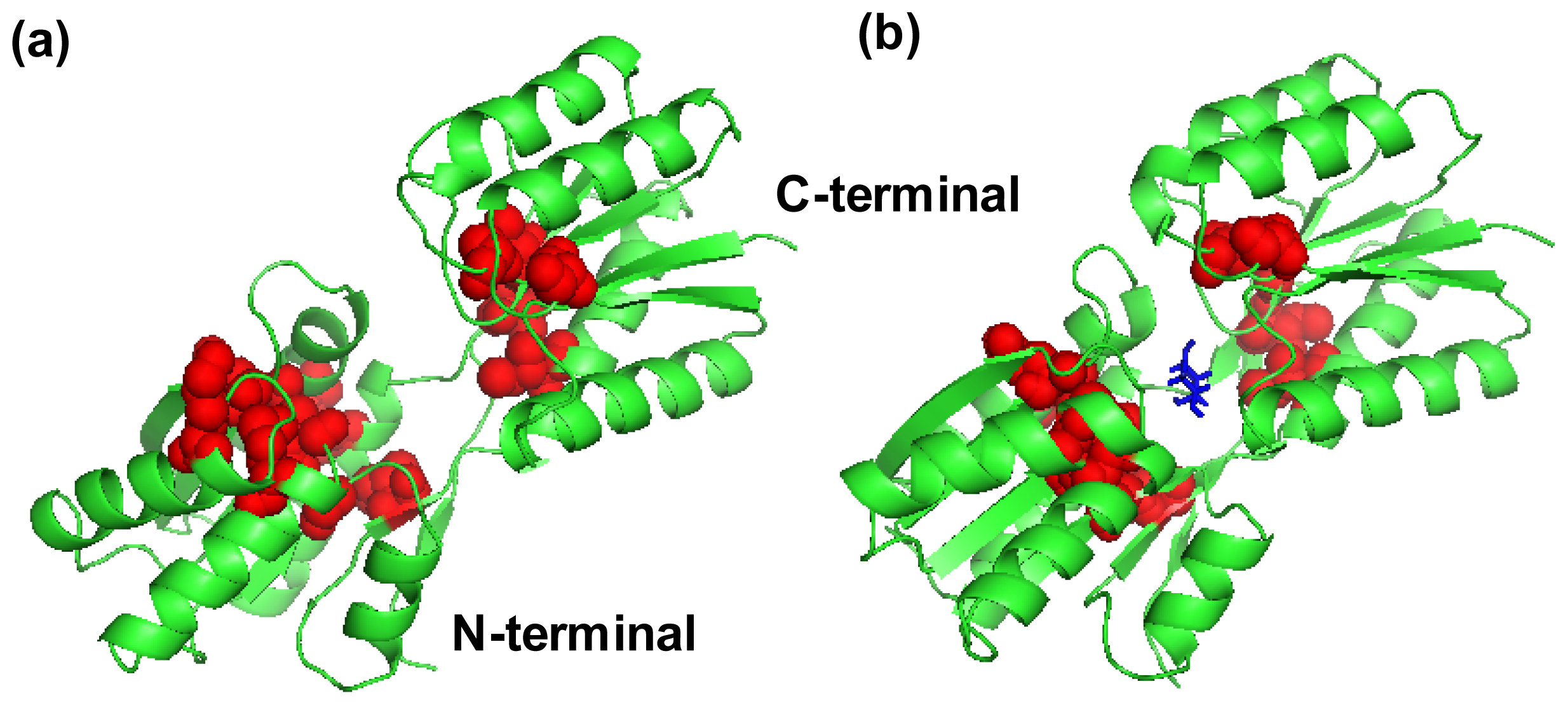
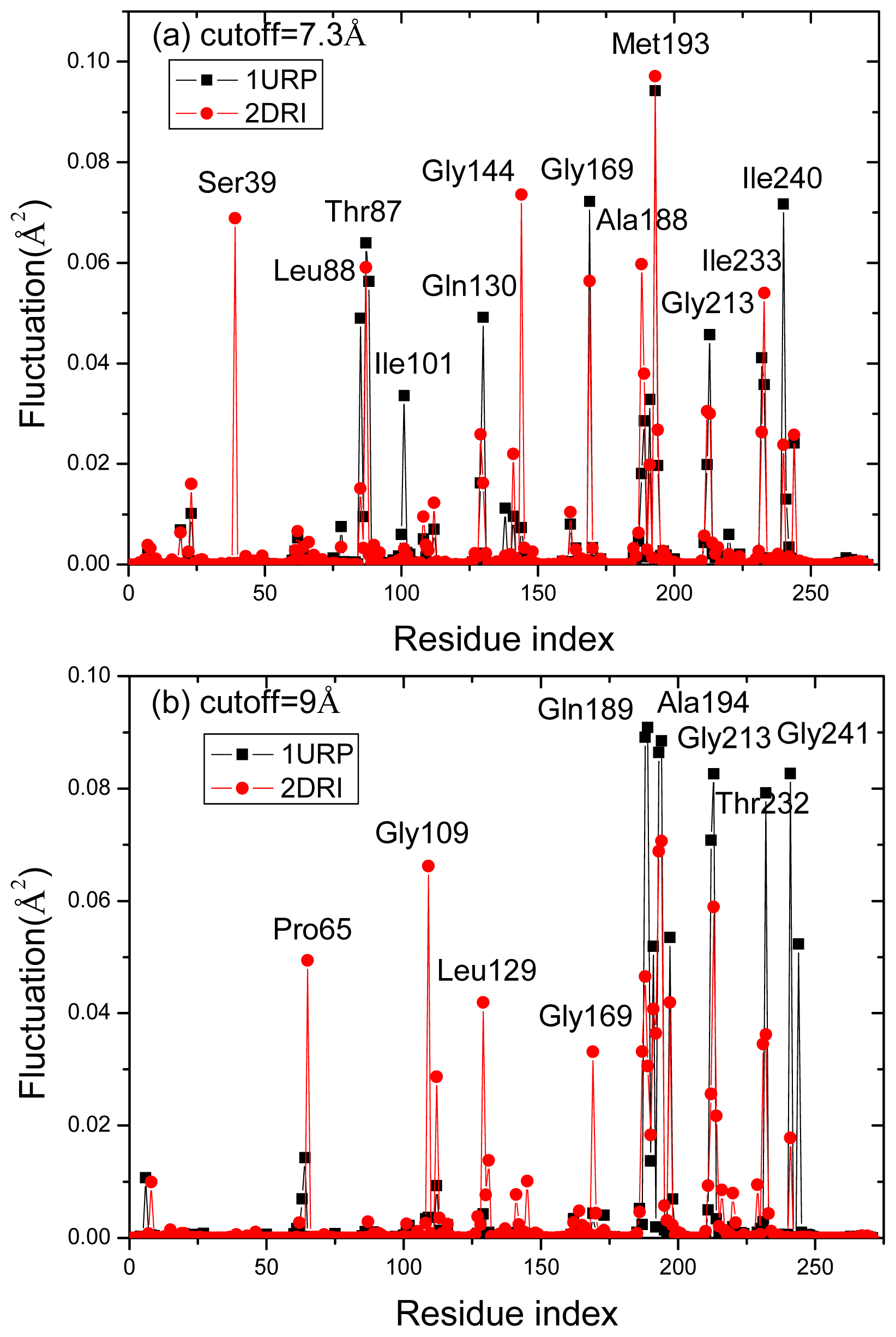
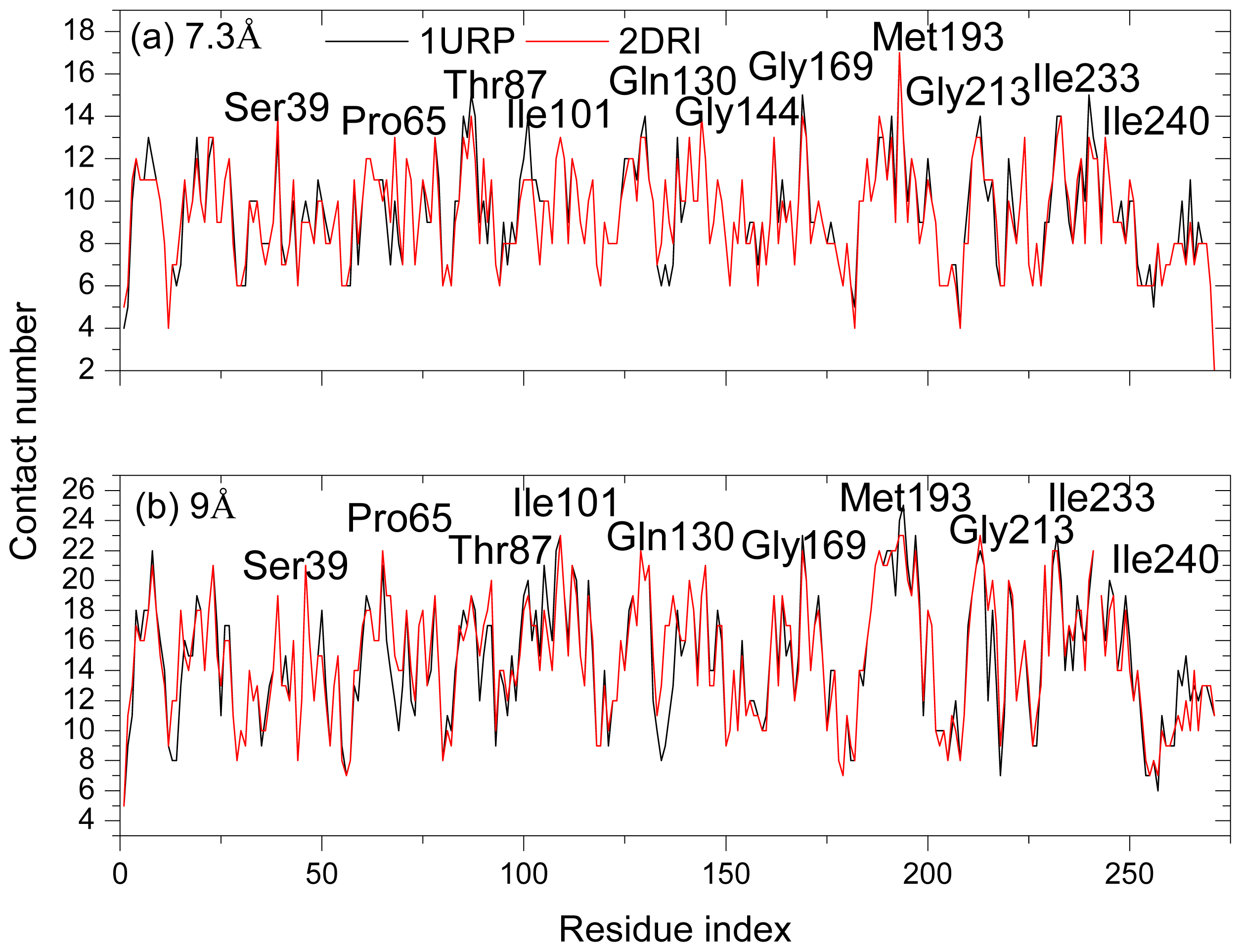
2.4. The Fast Mode of the Motion
3. Methods
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Anderson, C.; Zucker, F.; Steitz, T. Space-filling models of kinase clefts and conformation changes. Science 1979, 204, 375. [Google Scholar]
- Gerstein, M.; Lesk, A.M.; Chothia, C. Structural mechanisms for domain movements in proteins. Biochemistry 1994, 33, 6739–6749. [Google Scholar]
- Chen, X.; Schauder, S.; Potier, N.; van Dorsselaer, A.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 2002, 415, 545–549. [Google Scholar]
- Quiocho, F.; Ledvina, P. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: Variation of common themes. Mol. Microbiol 1996, 20, 17. [Google Scholar]
- Surin, B.; Rosenberg, H.; Cox, G. Phosphate-specific transport system of Escherichia coli: Nucleotide sequence and gene-polypeptide relationships. J. Bacteriol 1985, 161, 189–198. [Google Scholar]
- Tam, R.; Saier, M. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev 1993, 57, 320–346. [Google Scholar]
- Evenäs, J.; Tugarinov, V.; Skrynnikov, N.R.; Goto, N.K.; Muhandiram, R.; Kay, L.E. Ligand-induced structural changes to maltodextrin-binding protein as studied by solution NMR spectroscopy. J. Mol. Biol 2001, 309, 961–974. [Google Scholar]
- Magnusson, U.; Chaudhuri, B.N.; Ko, J.; Park, C.; Jones, T.A.; Mowbray, S.L. Hinge-bending motion of D-allose-binding protein from Escherichia coli three open conformations. J. Biol. Chem 2002, 277, 14077–14084. [Google Scholar]
- Bjorkman, A.J.; Mowbray, S.L. Multiple open forms of ribose-binding protein trace the path of its conformational change. J. Mol. Biol 1998, 279, 651–664. [Google Scholar]
- Hollenstein, K.; Frei, D.C.; Locher, K.P. Structure of an ABC transporter in complex with its binding protein. Nature 2007, 446, 213–216. [Google Scholar]
- Bjorkman, A.J.; Binnie, R.A.; Zhang, H.; Cole, L.B.; Hermodson, M.A.; Mowbray, S.L. Probing protein-protein interactions. The ribose-binding protein in bacterial transport and chemotaxis. J. Biol. Chem 1994, 269, 30206–30211. [Google Scholar]
- Mowbray, S.L.; Cole, L.B. 1.7 A X-ray structure of the periplasmic ribose receptor from Escherichia coli. J. Mol. Biol 1992, 225, 155–175. [Google Scholar]
- Luck, L.A.; Falke, J.J. Open conformation of a substrate-binding cleft: 19F NMR studies of cleft angle in the D-galactose chemosensory receptor. Biochemistry 1991, 30, 6484–6490. [Google Scholar]
- Tang, C.; Schwieters, C.D.; Clore, G.M. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature 2007, 449, 1078–1082. [Google Scholar]
- Loeffler, H.H.; Kitao, A. Collective dynamics of periplasmic glutamine binding protein upon domain closure. Biophys. J 2009, 97, 2541–2549. [Google Scholar]
- Ravindranathan, K.P.; Gallicchio, E.; Levy, R.M. Conformational equilibria and free energy profiles for the allosteric transition of the ribose-binding protein. J. Mol. Biol 2005, 353, 196–210. [Google Scholar]
- Stockner, T.; Vogel, H.J.; Tieleman, D.P. A salt-bridge motif involved in ligand binding and large-scale domain motions of the maltose-binding protein. Biophys. J 2005, 89, 3362–3371. [Google Scholar]
- Chang, G.; Guida, W.C.; Still, W.C. An internal coordinate monte-carlo method for searching conformational space. J. Am. Chem. Soc 1989, 111, 4379–4386. [Google Scholar]
- Su, J.G.; Jiao, X.; Sun, T.G.; Li, C.H.; Chen, W.Z.; Wang, C.X. Analysis of domain movements in glutamine-binding protein with simple models. Biophys. J 2007, 92, 1326–1335. [Google Scholar]
- Keskin, O. Comparison of full-atomic and coarse-grained models to examine the molecular fluctuations of c-AMP dependent protein kinase. J. Biomol. Struct. Dyn 2002, 20, 333–345. [Google Scholar]
- Vercillo, N.C.; Herald, K.J.; Fox, J.M.; Der, B.S.; Dattelbaum, J.D. Analysis of ligand binding to a ribose biosensor using site-directed mutagenesis and fluorescence spectroscopy. Protein Sci 2007, 16, 362–368. [Google Scholar]
- Shilton, B.H.; Flocco, M.M.; Nilsson, M.; Mowbray, S.L. Conformational changes of three periplasmic receptors for bacterial chemotaxis and transport: The maltose-, glucose/galactose-and ribose-binding proteins. J. Mol. Biol 1996, 264, 350–363. [Google Scholar]
- Zhang, Y.; Gardina, P.J.; Kuebler, A.S.; Der, B.S.; Dattelbaum, J.D. Model of maltose-binding protein/chemoreceptor complex supports intrasubunit signaling mechanism. Proc. Natl. Acad. Sci. USA 1999, 96, 939–944. [Google Scholar]
- Hayward, S.; Berendsen, H.J.C. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins Struct. Funct. Bioinf 1998, 30, 144–154. [Google Scholar]
- Haspel, N.; Moll, M.; Baker, M.L.; Chiu, W.; Kavraki, L.E. Tracing conformational changes in proteins. BMC Struct. Biol 2010, 10, S1. [Google Scholar]
- Mitsutake, A.; Sugita, Y.; Okamoto, Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Peptide Sci 2001, 60, 96–123. [Google Scholar]
- Lou, H.; Cukier, R.I. Molecular dynamics of apo-adenylate kinase: A distance replica exchange method for the free energy of conformational fluctuations. J. Phys. Chem. B 2006, 110, 24121–24137. [Google Scholar]
- Tirion, M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett 1996, 77, 1905–1908. [Google Scholar]
- Hinsen, K. Analysis of domain motions by approximate normal mode calculations. Proteins 1998, 33, 417–429. [Google Scholar]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des 1997, 2, 173–181. [Google Scholar]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian dynamics of folded proteins. Phys. Rev. Lett 1997, 79, 3090–3093. [Google Scholar]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J 2001, 80, 505–515. [Google Scholar]
- Micheletti, C.; Carloni, P.; Maritan, A. Accurate and efficient description of protein vibrational dynamics: Comparing molecular dynamics and Gaussian models. Proteins Struct. Funct. Bioinf 2004, 55, 635–645. [Google Scholar]
- Kundu, S.; Melton, J.S.; Sorensen, D.C.; Phillips, G.N. Dynamics of proteins in crystals: Comparison of experiment with simple models. Biophys. J 2002, 83, 723–732. [Google Scholar]
- Magnusson, U.; Salopek-Sondi, B.; Luck, L.A.; Mowbray, S.L. X-ray structures of the leucine-binding protein illustrate conformational changes and the basis of ligand specificity. J. Biol. Chem 2004, 279, 8747–8752. [Google Scholar]
- Heddle, J.; Scott, D.J.; Unzai, S.; Park, S.Y.; Tame, J.R. Crystal structures of the liganded and unliganded nickel-binding protein NikA from Escherichia coli. J. Biol. Chem 2003, 278, 50322–50329. [Google Scholar]
- Chaudhuri, B.N.; Ko, J.; Park, C.; Jones, T.A.; Mowbray, S.L. Structure of D-allose binding protein from Escherichia coli bound to D-allose at 1.8 A resolution. J. Mol. Biol 1999, 286, 1519–1531. [Google Scholar]
- Hsiao, C.D.; Sun, Y.J.; Rose, J.; Wang, B.C. The crystal structure of glutamine-binding protein from Escherichia coli. J. Mol. Biol 1996, 262, 225–242. [Google Scholar]
- Sun, Y.J.; Rose, J.; Wang, B.C.; Hsiao, C.D. The structure of glutamine-binding protein complexed with glutamine at 1.94 A resolution: Comparisons with other amino acid binding proteins. J. Mol. Biol 1998, 278, 219–229. [Google Scholar]
- Sharff, A.J.; Rodseth, L.E.; Spurlino, J.C.; Quiocho, F.A. Crystallographic evidence of a large ligand-induced hinge-twist motion between the 2 domains of the maltodextrin binding-protein involved in active-transport and chemotaxis. Biochemistry 1992, 31, 10657–10663. [Google Scholar]
- Quiocho, F.A.; Spurlino, J.C.; Rodseth, L.E. Extensive features of tight oligosaccharide binding revealed in high-resolution structures of the maltodextrin transport chemosensory receptor. Structure 1997, 5, 997–1015. [Google Scholar]
- Spurlino, J.C.; Lu, G.Y.; Quiocho, F.A. The 2.3-A resolution structure of the maltose- or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J. Biol. Chem 1991, 266, 5202–5219. [Google Scholar]
- Cuneo, M.J.; Beese, L.S.; Hellinga, H.W. Ligand-induced conformational changes in a thermophilic ribose-binding protein. BMC Struct. Biol 2008, 8, 50. [Google Scholar]
- Demirel, M.C.; Atilgan, A.L.I.R.; Jernigan, R.L.; Erman, B.; Bahar, I. Identification of kinetically hot residues in proteins. Protein Sci. 1998, 72522, 2522–2532. [Google Scholar]
- Vyas, N.K.; Vyas, M.N.; Quiocho, F.A. Sugar and signal-transducer binding sites of the Escherichia coli galactose chemoreceptor protein. Science 1988, 242, 1290–1295. [Google Scholar]
- Nishikawa, K.; Ooi, T.; Isogai, Y.; Saitô, N. Tertiary structure of proteins. I. Representation and computation of the conformations. J. Phys. Soc. Jpn 1972, 32, 1331–1337. [Google Scholar]
- Bahar, I.; Atilgan, A.R.; Demirel, M.C.; Erman, B. Vibrational dynamics of folded proteins: Significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett 1998, 80, 2733–2736. [Google Scholar]
- Rader, A.J.; Anderson, G.; Isin, B.; Khorana, H.G.; Bahar, I.; Klein-Seetharaman, J. Identification of core amino acids stabilizing rhodopsin. Proc. Natl. Acad. Sci. USA 2004, 101, 7246–7251. [Google Scholar]
- Haliloglu, T.; Keskin, O.; Ma, B.; Nussinov, R. How similar are protein folding and protein binding nuclei? Examination of vibrational motions of energy hot spots and conserved residues. Biophys. J 2005, 88, 1552. [Google Scholar]
- Keskin, O.; Ma, B.; Nussinov, R. Hot regions in protein-protein interactions: The organization and contribution of structurally conserved hot spot residues. J. Mol. Biol 2005, 345, 1281–1294. [Google Scholar]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 2005, 33, W299–W302. [Google Scholar]
- Binnie, R.A.; Zhang, H.; Mowbray, S.; Hermodson, M.A. Functional mapping of the surface of Escherichia coli ribose-binding protein: Mutations that affect chemotaxis and transport. Protein Sci 1992, 1, 1642–1651. [Google Scholar]
- Yang, L.W.; Liu, X.; Jursa, C.J.; Holliman, M.; Rader, A.J.; Karimi, H.A.; Bahar, I. iGNM: A database of protein functional motions based on Gaussian Network Model. Bioinformatics 2005, 21, 2978–2987. [Google Scholar]
- Eyal, E.; Yang, L.W.; Bahar, I. Anisotropic network model: Systematic evaluation and a new web interface. Bioinformatics 2006, 22, 2619–2627. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Periplasmic binding proteins | Pdb ID | Correlation | Reference | Resolution (Å) | |
|---|---|---|---|---|---|
| Leucine-binding protein | Apo | 1USG | 0.554 | Magnusson 2004 [35] | 1.53 |
| Complex | 1USI | 0.64 | 1.8 | ||
| Nickel-binding protein | Apo | 1UIU | 0.492 | Heddle 2003 [36] | 1.85 |
| Complex | 1UIV | 0.531 | 1.95 | ||
| Allose-binding protein | Apo | 1GUD | 0.69 | Chaudhuri 1999 [37] | 1.71 |
| Complex | 1RPJ | 0.594 | Magnusson 2002 [8] | 1.8 | |
| Glutamine-binding protein | Apo | 1GGG | 0.488 | Hsiao 1996 [38] | 2.3 |
| Complex | 1ANF | 0.581 | Sun 1998 [39] | 1.94 | |
| Maltose-binding protein | Apo | 1OMP | 0.653 | Sharff 1992 [40] | 1.8 |
| Complex | 1ANF | 0.594 | Quiocho 1997 [41] | 1.67 | |
| Ribose-binding protein | Apo | 1URP | 0.583 | Bjorkman 1998 [9] | 2.3 |
| Apo | 1BA2 | 0.669 | Bjorkman 1998 [9] | 2.1 | |
| Complex | 2DRI | 0.772 | Bjorkman 1994 [11] | 1.6 | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, H.Y.; Cao, Z.X.; Zhao, L.L.; Wang, J.H. Analysis of Conformational Motions and Residue Fluctuations for Escherichia coli Ribose-Binding Protein Revealed with Elastic Network Models. Int. J. Mol. Sci. 2013, 14, 10552-10569. https://doi.org/10.3390/ijms140510552
Li HY, Cao ZX, Zhao LL, Wang JH. Analysis of Conformational Motions and Residue Fluctuations for Escherichia coli Ribose-Binding Protein Revealed with Elastic Network Models. International Journal of Molecular Sciences. 2013; 14(5):10552-10569. https://doi.org/10.3390/ijms140510552
Chicago/Turabian StyleLi, Hai Yan, Zan Xia Cao, Li Ling Zhao, and Ji Hua Wang. 2013. "Analysis of Conformational Motions and Residue Fluctuations for Escherichia coli Ribose-Binding Protein Revealed with Elastic Network Models" International Journal of Molecular Sciences 14, no. 5: 10552-10569. https://doi.org/10.3390/ijms140510552