Dose-Dependent Protective Effect of Bisperoxovanadium against Acute Cerebral Ischemia in a Rat Model of Ischemia/Reperfusion Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
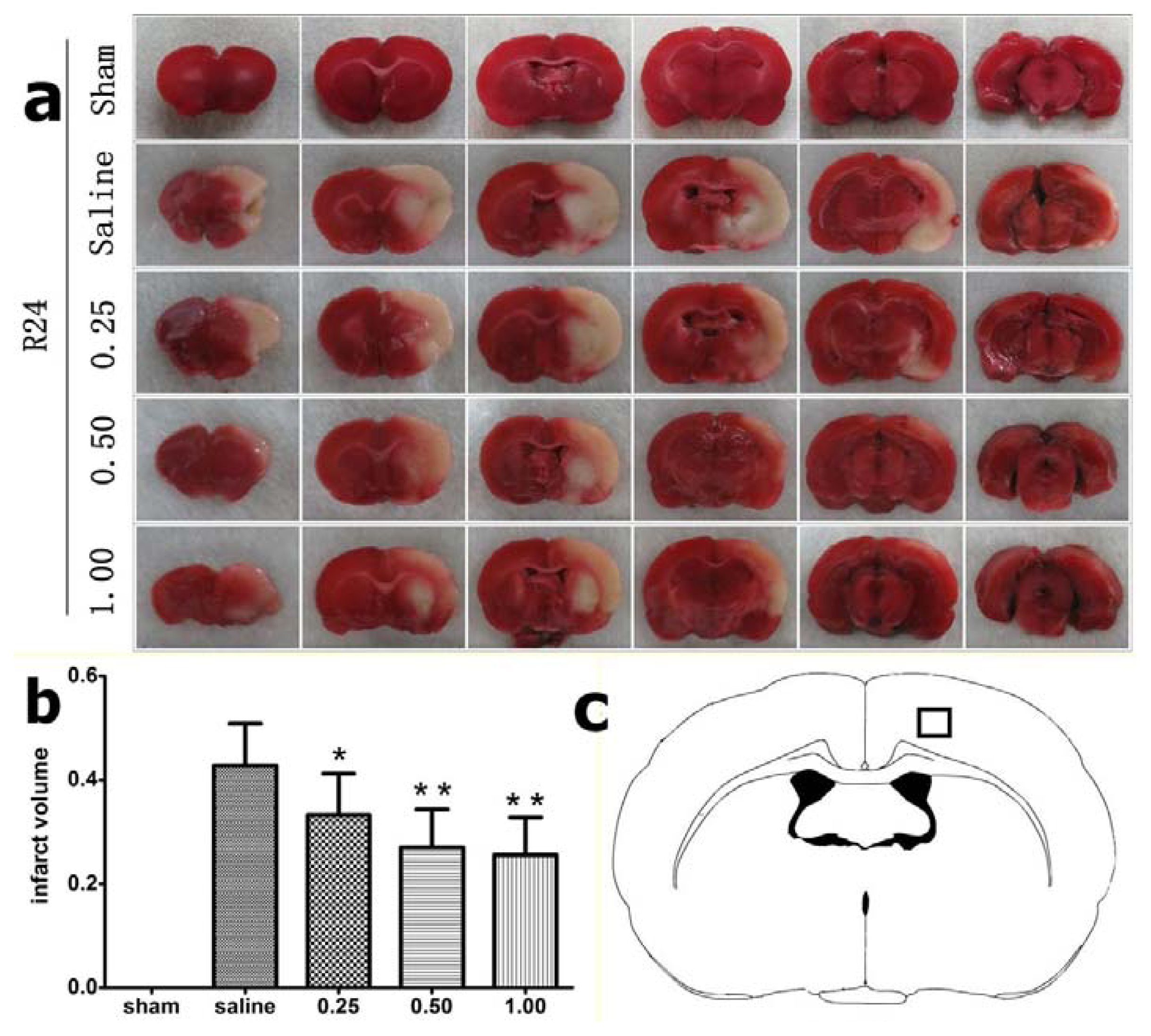
2.1. Effect of Bisperoxovanadium (bpV) on Infarct Volume
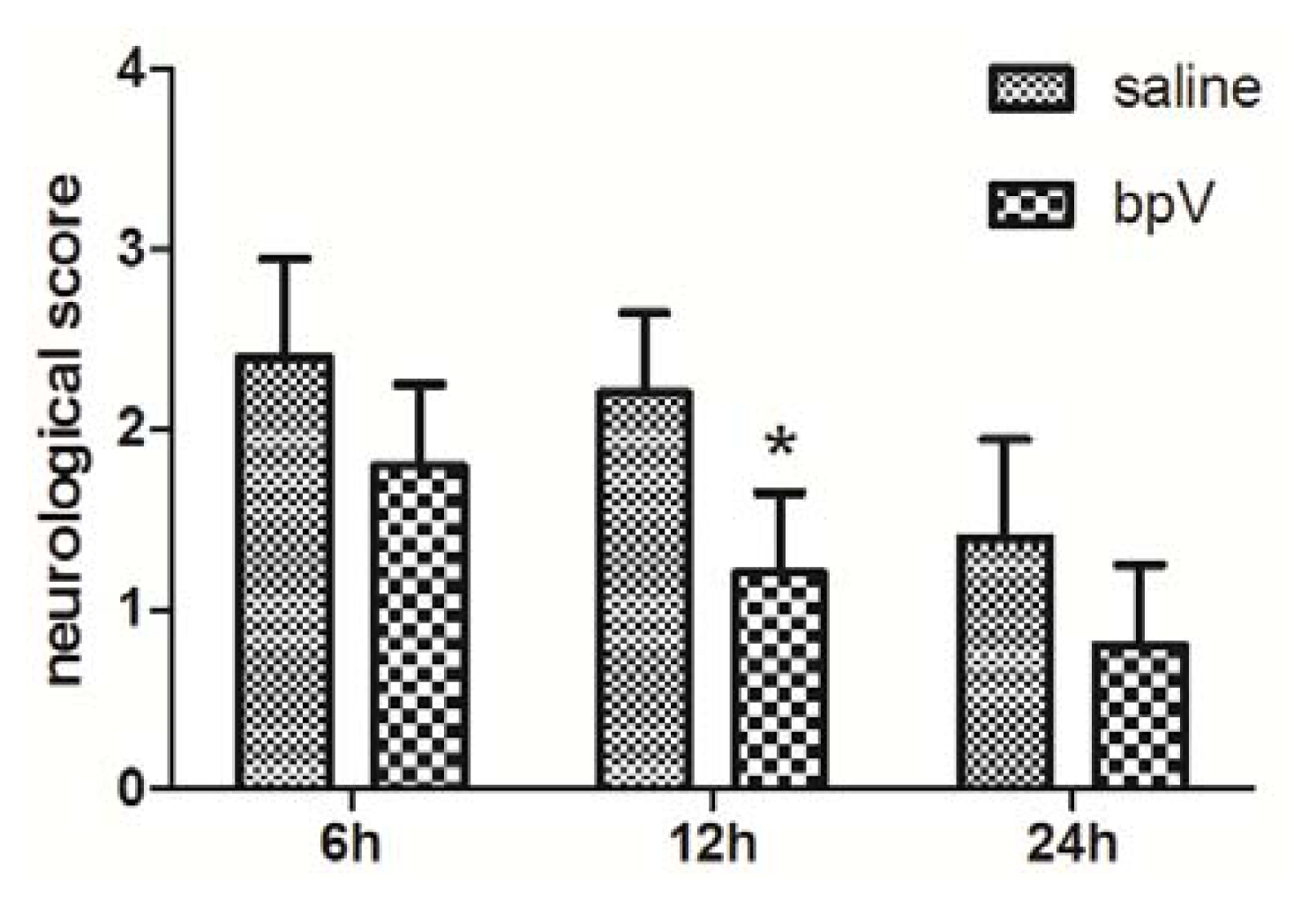
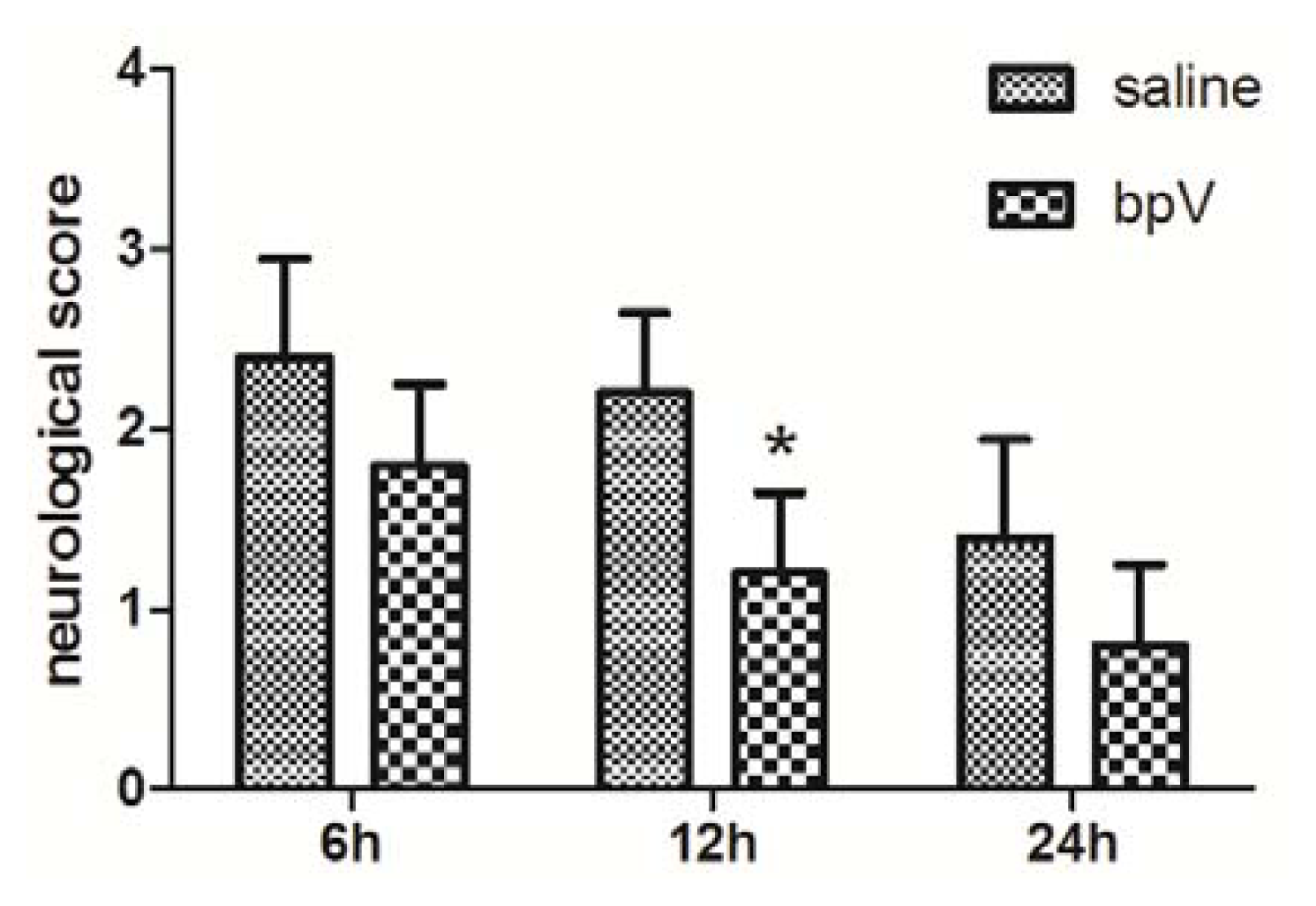
2.2. Effects of bpV on Neurological Deficits
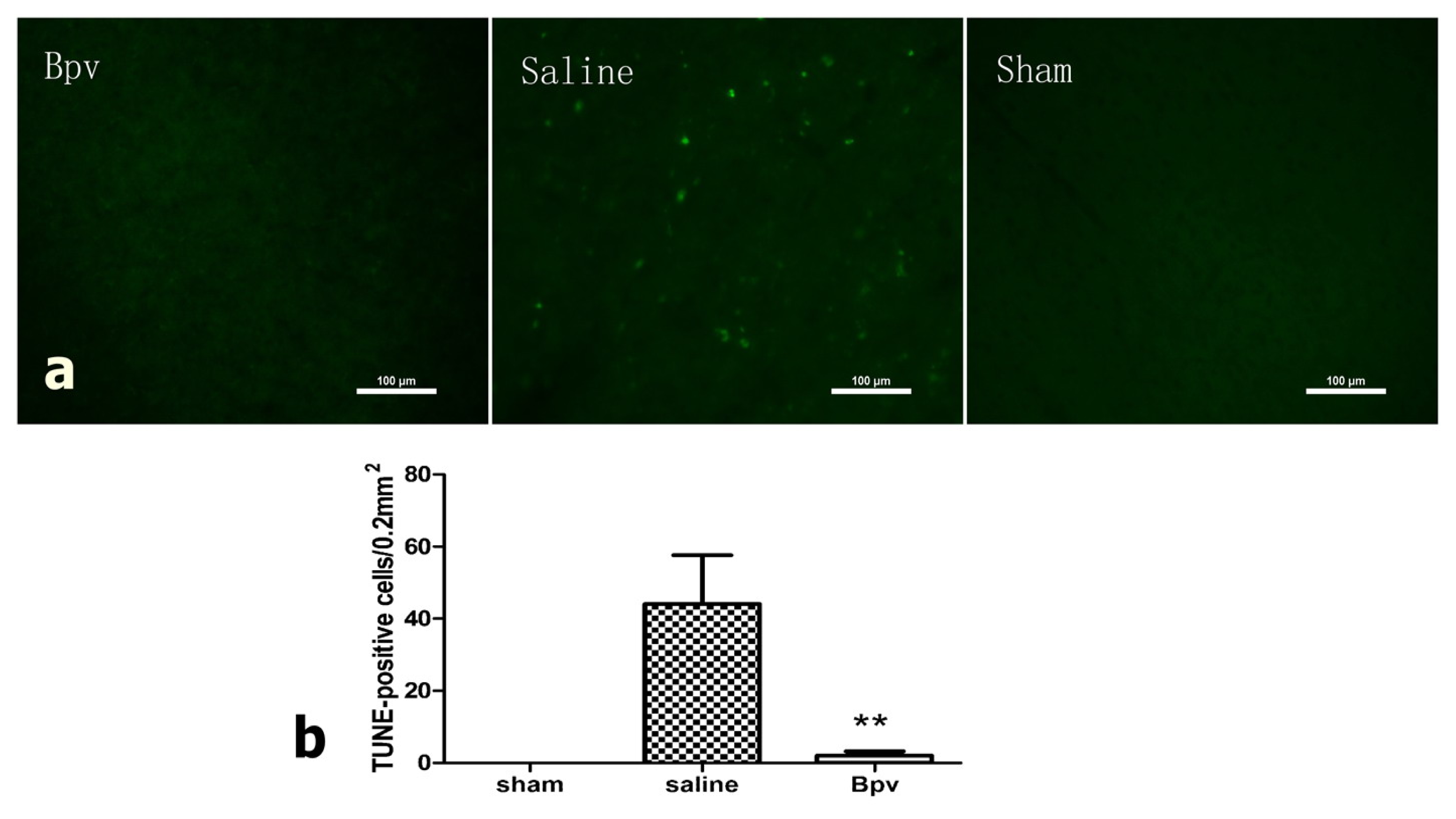
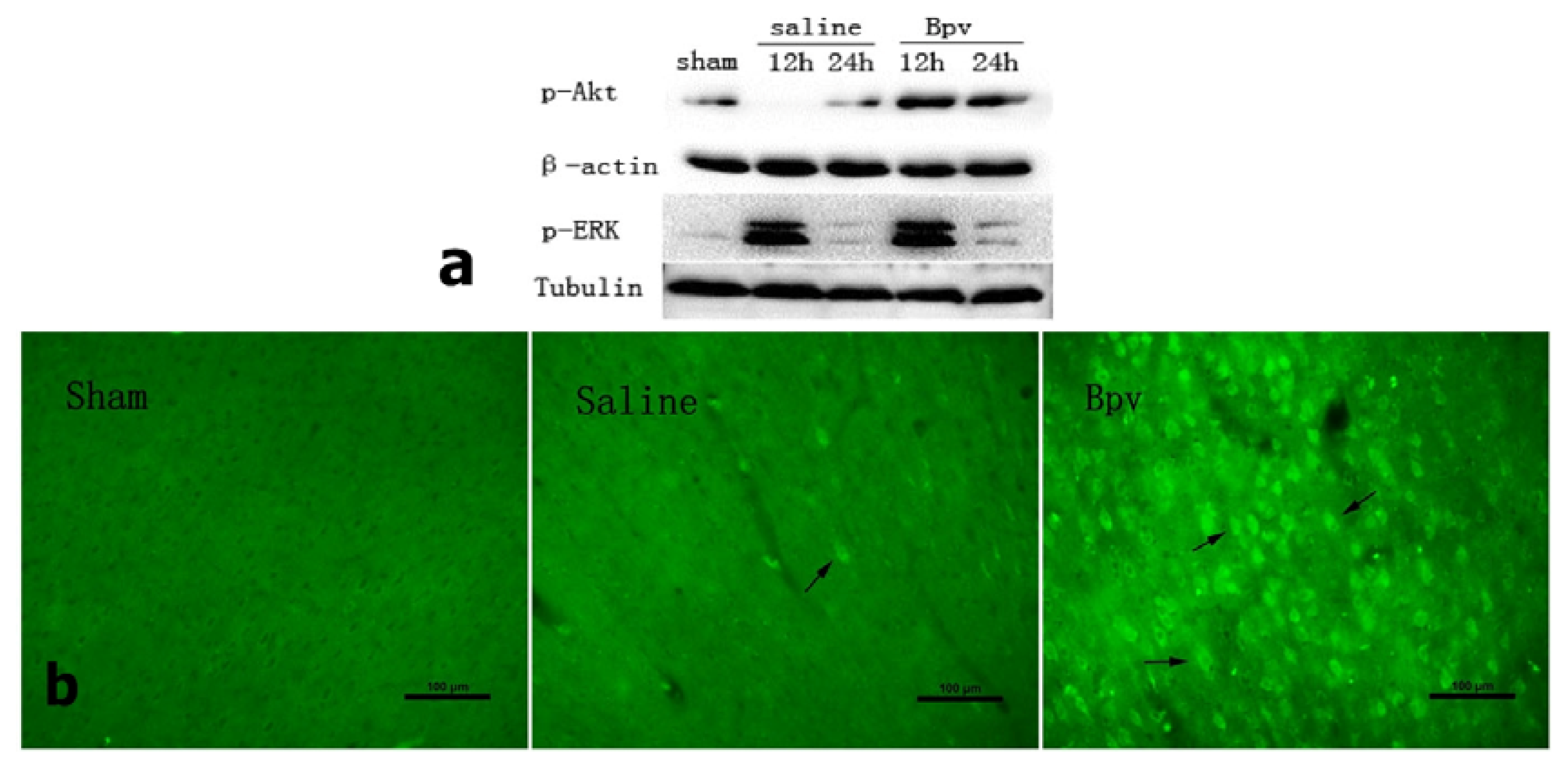
2.3. bpV Decreased Neuron Apoptosis Induced by Cerebral Ischemic/Reperfusion Injury
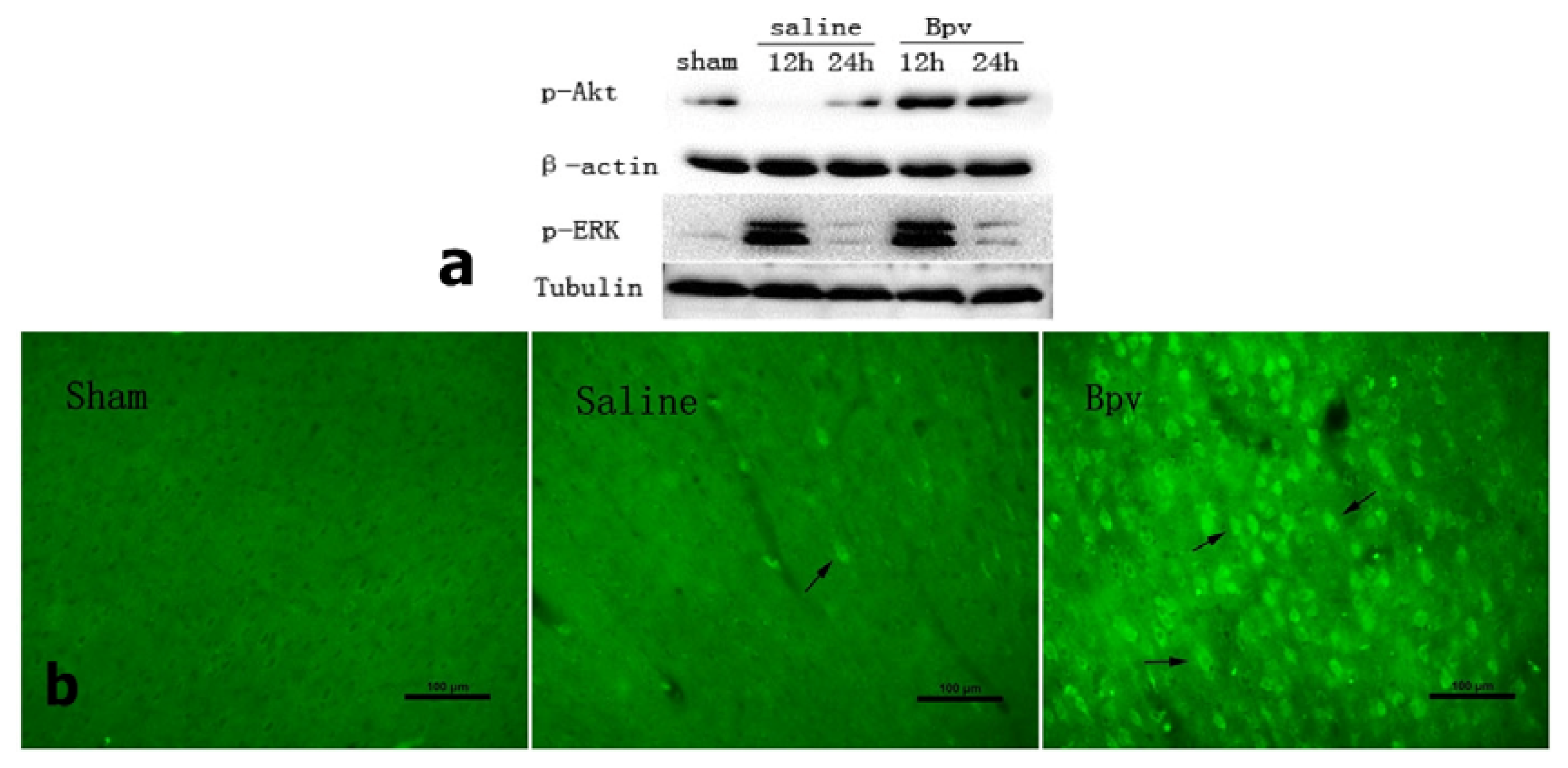
2.4. Effect of bpV on Phosphorylation of Akt (Ser473)
2.5. Discussion
3. Experimental Section
3.1. Ischemic Stroke Model
3.2. Bisperoxovanadium (bpV[HOpic]) Treatment
3.3. Infarct Volume Assessment
3.4. Neurological Deficit Evaluation
3.5. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL)
3.6. Western Blot Analysis
3.7. Immunohistochemistry
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Zaleska, M.M.; Mercado, M.L.; Chavez, J.; Feuerstein, G.Z.; Pangalos, M.N.; Wood, A. The development of stroke therapeutics: Promising mechanisms and translational challenges. Neuropharmacology 2009, 56, 329–341. [Google Scholar]
- Hacke, W.; Brott, T.; Caplan, L.; Meier, D.; Fieschi, C.; von Kummer, R.; Donnan, G.; Heiss, W.D.; Wahlgren, N.G.; Spranger, M.; et al. Thrombolysis in acute ischemic stroke: Controlled trials and clinical experience. Neurology 1999, 53, S3–S14. [Google Scholar]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar]
- Leslie, N.R.; Yang, X.; Downes, C.P.; Weijer, C.J. The regulation of cell migration by PTEN. Biochem. Soc. Trans 2005, 33, 1507–1508. [Google Scholar]
- Backman, S.; Stambolic, V.; Mak, T. PTEN function in mammalian cell size regulation. Curr. Opin. Neurobiol 2002, 12, 516–522. [Google Scholar]
- Ji, S.P.; Zhang, Y.; Van Cleemput, J.; Jiang, W.; Liao, M.; Li, L.; Wan, Q.; Backstrom, J.R.; Zhang, X. Disruption of PTEN coupling with 5-HT2C receptors suppresses behavioral responses induced by drugs of abuse. Nat. Med 2006, 12, 324–329. [Google Scholar]
- Kwon, C.H.; Zhu, X.; Zhang, J.; Knoop, L.L.; Tharp, R.; Smeyne, R.J.; Eberhart, C.G.; Burger, P.C.; Baker, S.J. Pten regulates neuronal soma size: A mouse model of Lhermitte-Duclos disease. Nat. Genet 2001, 29, 404–411. [Google Scholar]
- Omori, N.; Jin, G.; Li, F.; Zhang, W.R.; Wang, S.J.; Hamakawa, Y.; Nagano, I.; Manabe, Y.; Shoji, M.; Abe, K. Enhanced phosphorylation of PTEN in rat brain after transient middle cerebral artery occlusion. Brain Res 2002, 954, 317–322. [Google Scholar]
- Walker, C.L.; Walker, M.J.; Liu, N.K.; Risberg, E.C.; Gao, X.; Chen, J.; Xu, X.M. Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury. PLoS One 2012, 7, e30012. [Google Scholar]
- Huang, X.; Zhang, H.; Yang, J.; Wu, J.; McMahon, J.; Lin, Y.; Cao, Z.; Gruenthal, M.; Huang, Y. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol. Dis 2010, 40, 193–199. [Google Scholar]
- Choi, Y.C.; Lee, J.H.; Hong, K.W.; Lee, K.S. 17 Beta-estradiol prevents focal cerebral ischemic damages via activation of Akt and CREB in association with reduced PTEN phosphorylation in rats. Fundam. Clin. Pharmacol 2004, 18, 547–557. [Google Scholar]
- Schmid, A.C.; Byrne, R.D.; Vilar, R.; Woscholski, R. Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett 2004, 566, 35–38. [Google Scholar]
- Shackelford, D.A.; Yeh, R.Y. Modulation of ERK and JNK activity by transient forebrain ischemia in rats. J. Neurosci. Res 2006, 83, 476–488. [Google Scholar]
- Wahl, F.; Allix, M.; Plotkine, M.; Boulu, R.G. Neurological and behavioral outcomes of focal cerebral ischemia in rats. Stroke 1992, 23, 267–272. [Google Scholar]
- Yang, Y.; Li, Q.; Miyashita, H.; Howlett, W.; Siddiqui, M.; Shuaib, A. Usefulness of postischemic thrombolysis with or without neuroprotection in a focal embolic model of cerebral ischemia. J. Neurosurg 2000, 92, 841–847. [Google Scholar]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar]
- Hetman, M.; Cavanaugh, J.E.; Kimelman, D.; Xia, Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J. Neurosci 2000, 20, 2567–2574. [Google Scholar]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar]
- Liu, K.; Lu, Y.; Lee, J.K.; Samara, R.; Willenberg, R.; Sears-Kraxberger, I.; Tedeschi, A.; Park, K.K.; Jin, D.; Cai, B.; et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci 2010, 13, 1075–1081. [Google Scholar]
- Gary, D.S.; Mattson, M.P. PTEN regulates Akt kinase activity in hippocampal neurons and increases their sensitivity to glutamate and apoptosis. Neuromol. Med 2002, 2, 261–269. [Google Scholar]
- Ruan, H.; Li, J.; Ren, S.; Gao, J.; Li, G.; Kim, R.; Wu, H.; Wang, Y. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J. Mol. Cell. Cardiol 2009, 46, 193–200. [Google Scholar]
- Liao, J.; Marumoto, T.; Yamaguchi, S.; Okano, S.; Takeda, N.; Sakamoto, C.; Kawano, H.; Nii, T.; Miyamato, S.; Nagai, Y.; et al. Inhibition of PTEN Tumor Suppressor Promotes the Generation of Induced Pluripotent Stem Cells. Mol. Ther. 2013. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Park, K.K.; Belin, S.; Wang, D.; Lu, T.; Chen, G.; Zhang, K.; Yeung, C.; Feng, G.; Yankner, B.A.; He, Z. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 2011, 480, 372–375. [Google Scholar] [Green Version]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar]
- Du, W.; Huang, J.; Yao, H.; Zhou, K.; Duan, B.; Wang, Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. J. Clin. Invest 2010, 120, 3480–3492. [Google Scholar]
- Swanson, R.A.; Morton, M.T.; Tsao-Wu, G.; Savalos, R.A.; Davidson, C.; Sharp, F.R. A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab. 1990, 10, 290–293. [Google Scholar]
- Saito, A.; Hayashi, T.; Okuno, S.; Ferrand-Drake, M.; Chan, P.H. Overexpression of copper/zinc superoxide dismutase in transgenic mice protects against neuronal cell death after transient focal ischemia by blocking activation of the Bad cell death signaling pathway. J. Neurosci 2003, 23, 1710–1718. [Google Scholar]
- Ashwal, S.; Tone, B.; Tian, H.R.; Cole, D.J.; Pearce, W.J. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke 1998, 29, 1037–1046, ; discussion 1047.. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, J.-Y.; Ding, J.; Yuan, F.; Chen, H.; Chen, S.-W.; Tian, H.-L. Dose-Dependent Protective Effect of Bisperoxovanadium against Acute Cerebral Ischemia in a Rat Model of Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2013, 14, 12013-12022. https://doi.org/10.3390/ijms140612013
Guo J-Y, Ding J, Yuan F, Chen H, Chen S-W, Tian H-L. Dose-Dependent Protective Effect of Bisperoxovanadium against Acute Cerebral Ischemia in a Rat Model of Ischemia/Reperfusion Injury. International Journal of Molecular Sciences. 2013; 14(6):12013-12022. https://doi.org/10.3390/ijms140612013
Chicago/Turabian StyleGuo, Jian-Yi, Jun Ding, Fang Yuan, Hao Chen, Shi-Wen Chen, and Heng-Li Tian. 2013. "Dose-Dependent Protective Effect of Bisperoxovanadium against Acute Cerebral Ischemia in a Rat Model of Ischemia/Reperfusion Injury" International Journal of Molecular Sciences 14, no. 6: 12013-12022. https://doi.org/10.3390/ijms140612013
APA StyleGuo, J.-Y., Ding, J., Yuan, F., Chen, H., Chen, S.-W., & Tian, H.-L. (2013). Dose-Dependent Protective Effect of Bisperoxovanadium against Acute Cerebral Ischemia in a Rat Model of Ischemia/Reperfusion Injury. International Journal of Molecular Sciences, 14(6), 12013-12022. https://doi.org/10.3390/ijms140612013