Protein Folding and Aggregation into Amyloid: The Interference by Natural Phenolic Compounds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
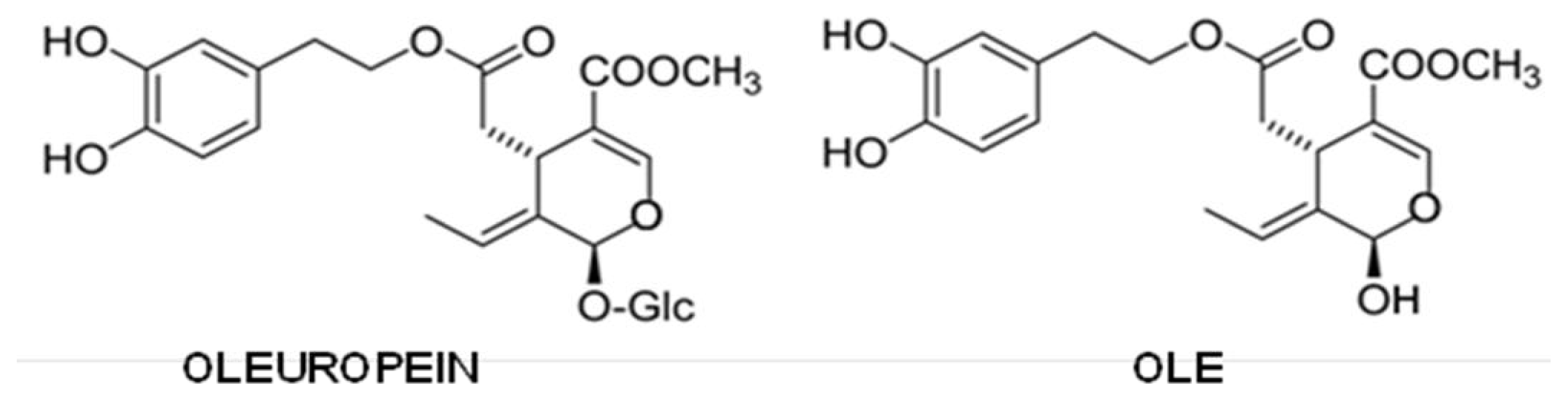{kind=link}
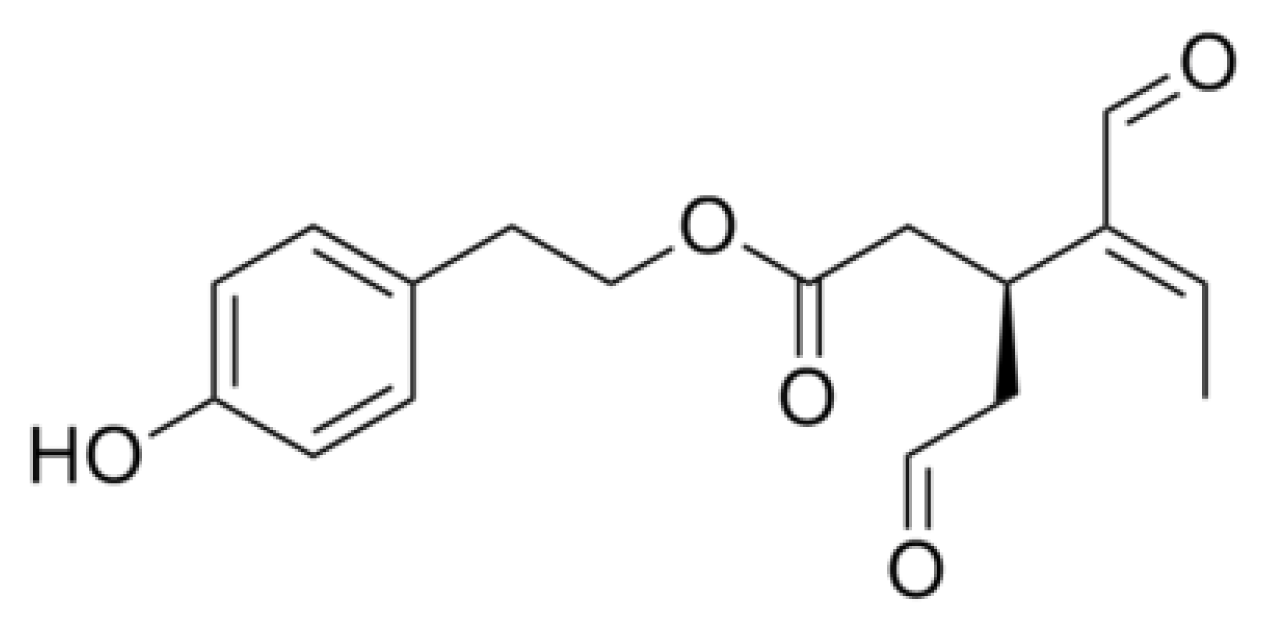{kind=link}
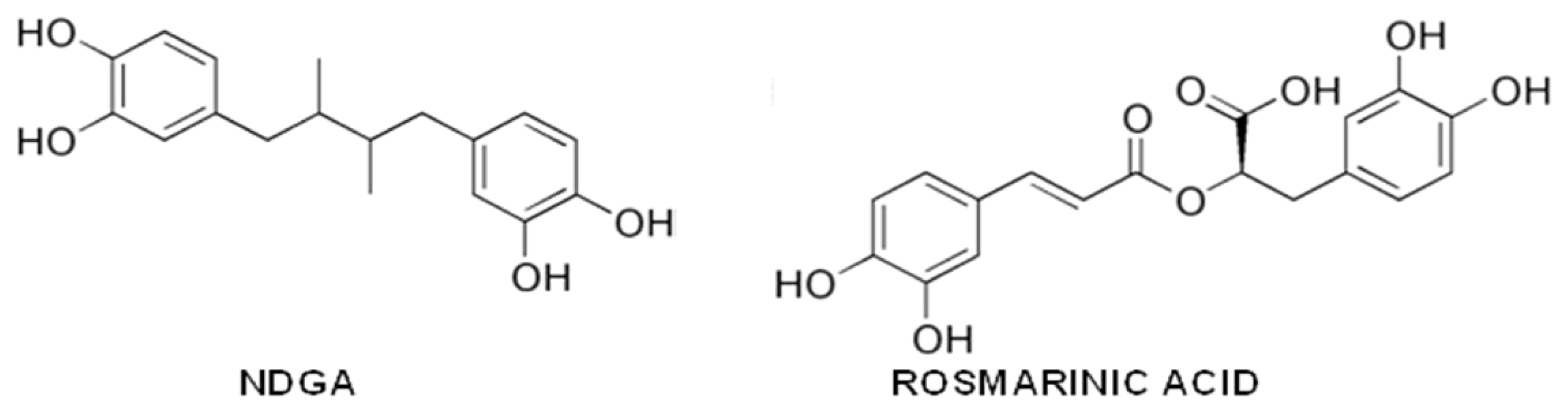{kind=link}
Abstract
:1. Introduction
2. Amyloid Aggregation: Overview and Mechanisms
3. Amyloid Aggregates: Mechanisms of Cytotoxicity
4. Amyloid Aggregation Inhibitors: An Overview
5. The Anti-Amyloid Properties of Natural Phenols and Polyphenols
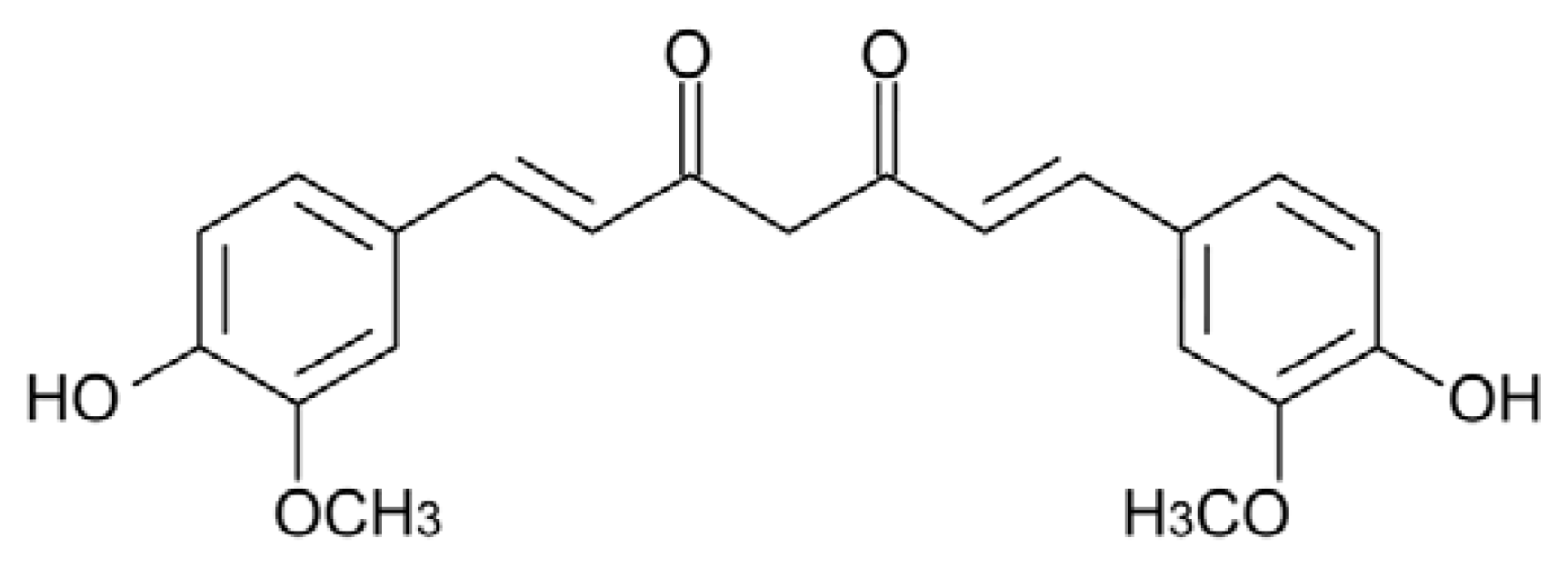
5.1. Curcumin
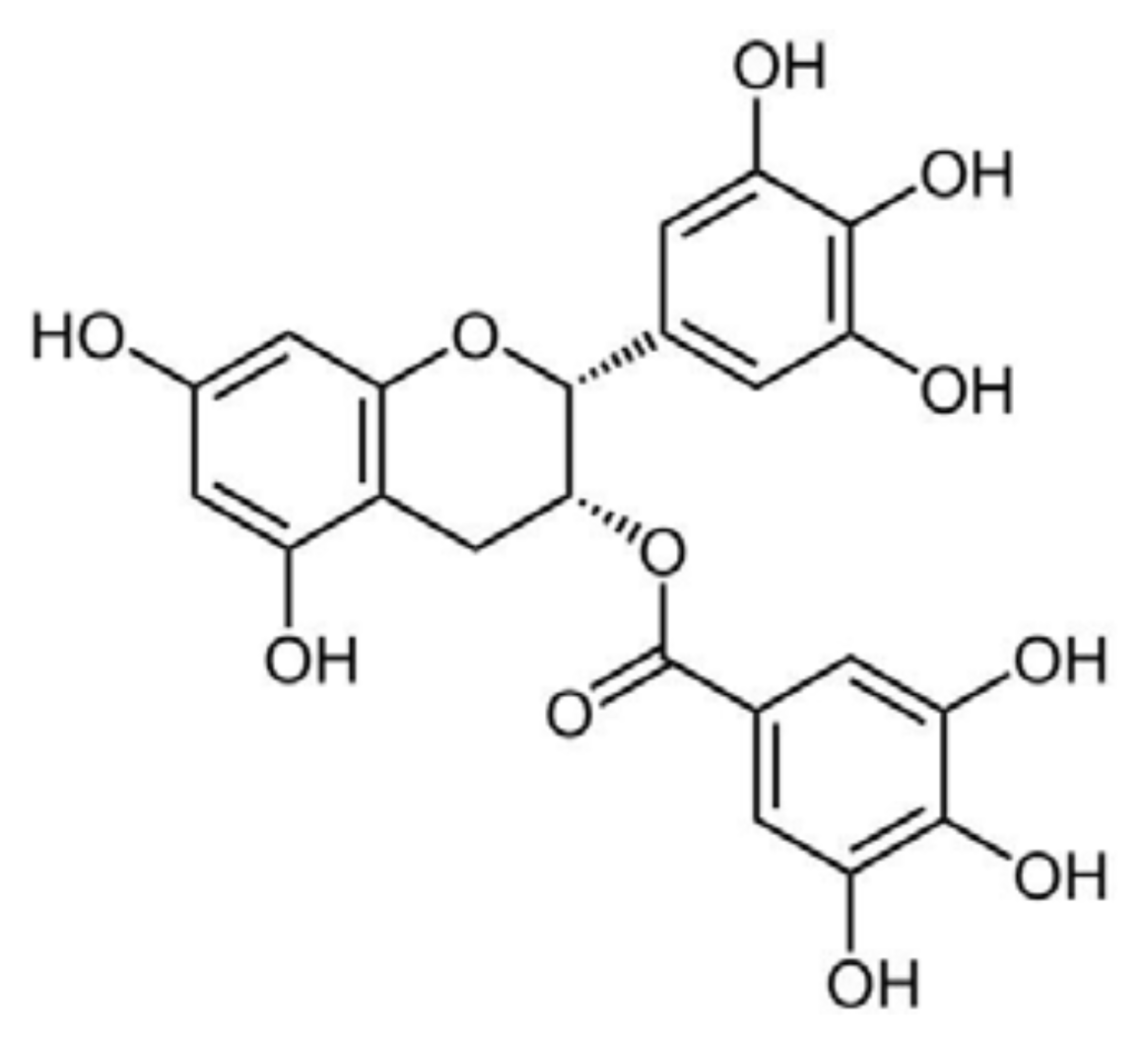
5.2. Epigallocatechin-Gallate
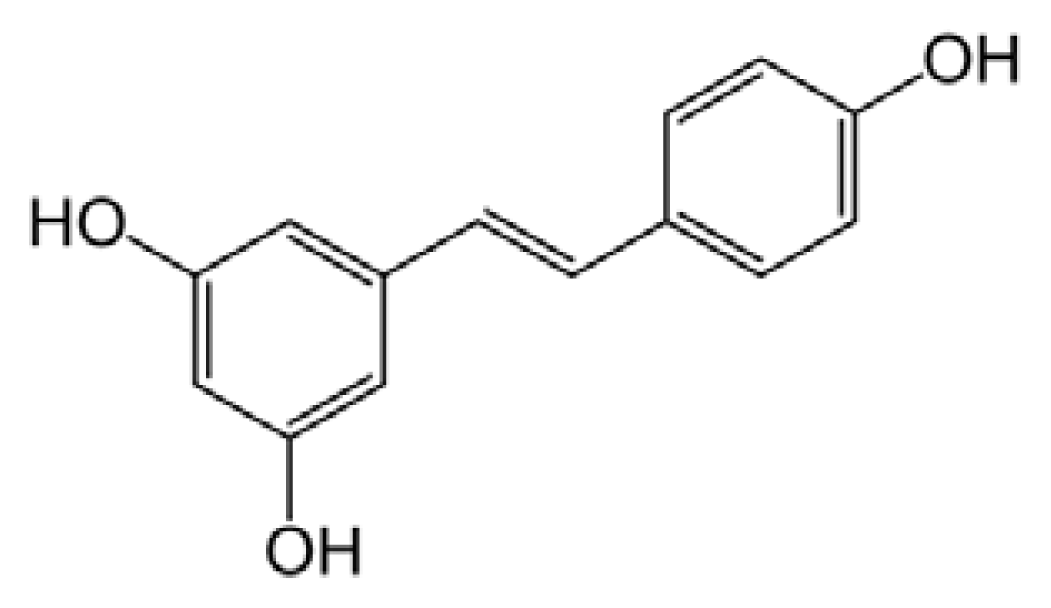
5.3. Resveratrol
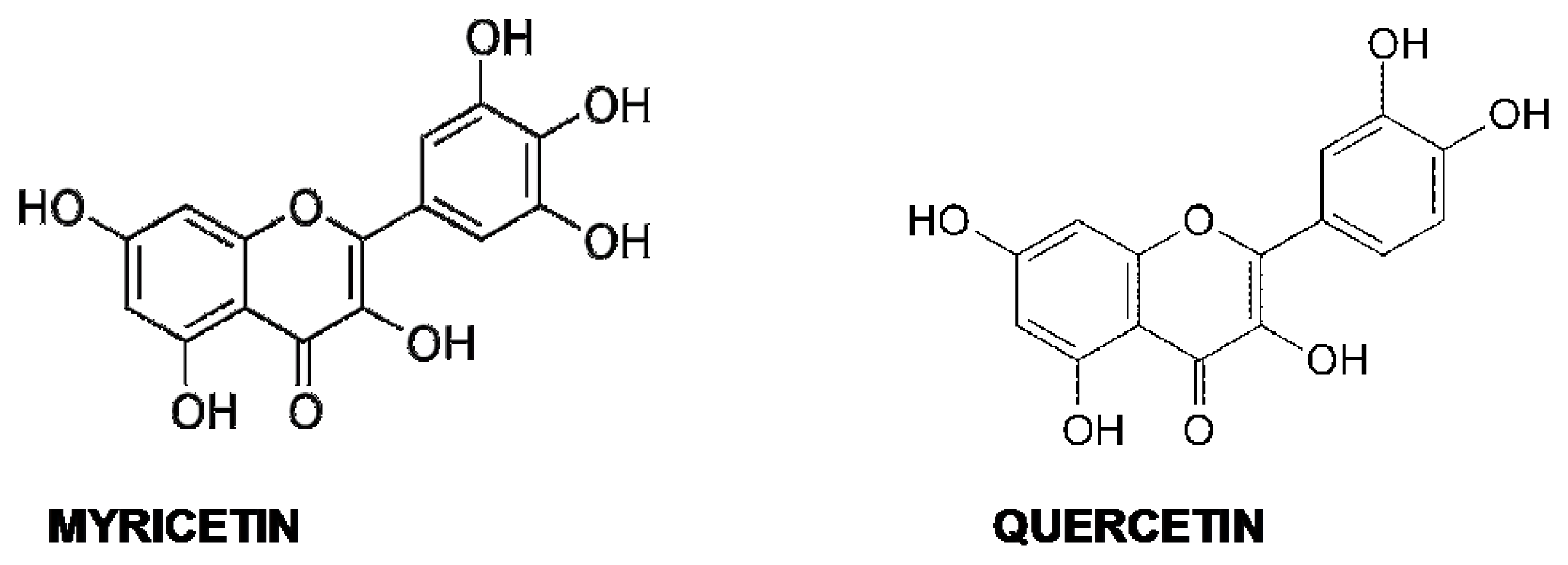
5.4. Quercetin and Myricetin
5.5. Olive Oil Phenols
5.6. Other Phenols
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Levinthal, C. Are there pathways for protein folding? J. Chem. Phys 1968, 85, 44–45. [Google Scholar]
- Radford, S.E.; Dobson, C.M. From computer simulations to human disease: Emerging themes in protein folding. Cell 1999, 97, 291–298. [Google Scholar]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med 2003, 81, 678–699. [Google Scholar]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem 2006, 75, 333–366. [Google Scholar]
- Thomas, P.J.; Qu, B.-H.; Pedersen, P.L. Defective protein folding as a basis of human disease. Trends Biochem. Sci 1995, 20, 456–459. [Google Scholar]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar]
- Serpell, L.C.; Sunde, M.; Benson, M.D.; Tennent, G.A.; Pepys, M.B.; Fraser, P.E. The protofilament substructure of amyloid fibrils. J. Mol. Biol 2000, 300, 1033–1039. [Google Scholar]
- Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Partitioning conformational intermediates between competing refolding and aggregation pathways: Insights into transthyretin amyloid disease. Biochemistry 2005, 44, 16612–16623. [Google Scholar]
- Jahn, T.T.; Radford, S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys 2008, 469, 100–117. [Google Scholar]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.I.; Shammas, S.L.; Wuadby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc 2011, 133, 14160–14163. [Google Scholar]
- Gama Sosa, M.A.; de Gasperi, R.; Elder, G.A. Modeling human neurodegenerative diseases in transgenic systems. Hum. Genet 2012, 131, 535–563. [Google Scholar]
- Clarke, G.; Collins, R.A.; Leavitt, B.R.; Andrews, D.F.; Hayden, M.R.; Lumsden, C.J.; McInnes, R.R. A one-hit model of cell death in inherited neuronal degeneration. Nature 2000, 406, 195–199. [Google Scholar]
- Perutz, M.F.; Windle, A.H. Cause of neuronal death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature 2001, 412, 143–144. [Google Scholar]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible nonfibrillar ligands derived from Aβ are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 93, 6448–6453. [Google Scholar]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem 1999, 274, 25945–25952. [Google Scholar]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar]
- Conway, K.A.; Lee, S.-J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of oligomerization not fibrillization is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease. Implication for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar]
- Reixach, N.; Deechingkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822. [Google Scholar]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar]
- Hirakura, Y.; Kagan, B.L. Pore formation by beta-2-microglobulin: A mechanism for the pathogenesis of dialysis-associated amyloidosis. Amyloid 2001, 8, 94–100. [Google Scholar]
- Van Horssen, J.; Wilhelmus, M.M.; Heljasvaara, R.; Pihlajaniemi, T.; Wesseling, P.; de Waal, R.M.; Verbeek, M.M. Collagen XVIII: A novel heparan sulfate proteoglycan associated with vascular amyloid depositions and senile plaques in Alzheimer’s disease brains. Brain Pathol 2002, 12, 456–462. [Google Scholar]
- Diaz-Nido, J.; Wandosell, F.; Avila, J. Glycosaminoglycans and beta-amyloid, prion and tau peptides in neurodegenerative diseases. Peptides 2002, 23, 1323–1332. [Google Scholar]
- Pepys, M.B.; Herbert, J.; Hutchinson, W.L.; Tennent, G.A.; Lachmann, H.J.; Gallimore, J.R.; Lovat, L.B.; Bartfal, T.; Alanine, A.; Hertel, C.; et al. Targeted pharmaceutical depletion of serum amyloid P component for treatment of human amyloidosis. Nature 2002, 417, 254–259. [Google Scholar]
- Walsh, D.M.; Selkoe, D.J. Oligomers on the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein Peptide Lett 2004, 11, 1–16. [Google Scholar]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β specifically disrupt cognitive function. Nature Neurosci 2005, 8, 79–84. [Google Scholar]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar]
- Koffie, R.M.; Meyer-Luehmann, M.; Hashimoto, T.; Adams, K.W.; Mielke, M.L.; Garcia-Alloza, M.; Micheva, K.D.; Smith, S.J.; Kim, M.L.; Lee, V.M.; et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. USA 2009, 106, 4012–4017. [Google Scholar]
- Nekooki-Machida, Y.; Kurosawa, M.; Nukina, N.; Ito, K.; Oda, T.; Tanaka, M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9679–9684. [Google Scholar]
- Gharibyan, A.L.; Zamotin, V.; Yanamandra, K.; Moskaleva, O.S.; Margulis, B.A.; Kostanyan, I.A.; Morozova-Roche, L.A. Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J. Mol. Biol 2007, 365, 1337–1349. [Google Scholar]
- Novitskaya, V.; Bocharova, O.V.; Bronstein, I.; Baskakov, I.V. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J. Biol. Chem 2006, 281, 13828–13836. [Google Scholar]
- Bucciantini, M.; Nosi, D.; Forzan, M.; Russo, E.; Calamai, M.; Pieri, L.; Formigli, L.; Quercioli, F.; Soria, S.; Pavone, F.; et al. Toxic effects of amyloid fibrils on cell membranes: The importance of ganglioside GM1. FASEB J 2012, 26, 818–831. [Google Scholar]
- Martins, I.C.; Kuperstein, I.; Wilkinson, H.; Maes, E.; Vambrabant, M.; Jonckheere, W.; van Gelder, P.; Hartmann, D.; D’Hooge, R.; de Strooper, B.; et al. Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J 2008, 27, 224–233. [Google Scholar]
- Xue, W.-F.; Hellewell, A.L.; Gosal, W.S.; Homans, S.W.; Hewitt, E.W.; Radford, S.E. Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem 2009, 284, 34272–34282. [Google Scholar]
- Klyubin, I.; Walsh, D.M.; Lemere, C.A.; Cullen, V.K.; Shankar, G.M.; Betts, V.; Spooner, E.T.; Jiang, L.; Amwyl, R.; Selkoe, D.J.; et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat. Med 2005, 11, 556–561. [Google Scholar]
- Chafekar, F.M.; Hoozemans, J.J.; Zwart, R.; Baas, F.; Scheper, W. Abeta 1–42 induces mild endoplasmic reticulum stress in an aggregation state-dependent manner. Antiox. Red. Signal 2007, 9, 2245–2254. [Google Scholar]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J 2005, 19, 2030–2041. [Google Scholar]
- Eikelemboom, R.; Bate, C.; van Gool, W.A.; Hoozemans, J.J.; Rozemuller, J.M.; Veerhuis, R.; Williams, A. Neuroinflammation in Alzheimer’s disease and prion disease. Glia 2002, 40, 232–239. [Google Scholar]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.E.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout in a mouse model of Alzheimer’s disease. Nat. Neurosci 2010, 13, 411–413. [Google Scholar]
- Chafekar, S.M.; Baas, F.; Scheper, W. Oligomer-specific Aβ toxicity in cell models is mediated by selective uptake. Biochim. Biophys. Acta 2008, 1782, 523–531. [Google Scholar]
- Dickson, D.W. Correlation of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging 1995, 16, 285–298. [Google Scholar]
- Powers, E.T.; Balch, W.E. Diversity in the origins of proteostasis networks—A driver for protein function in evolution. Nat. Rev. Mol. Cell Biol 2013, 14, 237–248. [Google Scholar]
- Vivekanandan, S.; Brender, J.R.; Lee, S.Y.; Ramamoorthy, A. A partially folded structure of Amyloid-Beta (1–40) in an aqueous environment. Biochem. Biophys. Res. Commun 2011, 411, 312–316. [Google Scholar]
- Yonemoto, I.T.; Kroon, G.J.; Dyson, H.J.; Balch, W.E.; Kelly, J.W. Amylin proprotein proessing generates progressively more amyloidogenica peptides that initially sample the helical state. Biochemistry 2008, 47, 9900–9910. [Google Scholar]
- Colon, W.; Kelly, J.W. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 1992, 31, 8654–8660. [Google Scholar]
- Chiti, F.; Webster, P.; Taddei, N.; Clark, A.; Stefani, M.; Ramponi, G.; Dobson, C.M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA 1999, 96, 3590–3594. [Google Scholar]
- McParland, V.J.; Kad, N.M.; Kalverda, A.P.; Brown, A.; Kirwin-Jones, P.; Hunter, M.G.; Sunde, M.; Radford, S.E. Partially unfolded states of beta(2)-microglobulin and amyloid formation in vitro. Biochemistry 2000, 39, 8735–8746. [Google Scholar]
- Harper, J.D.; Lansbury, P.T. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem 1997, 66, 385–407. [Google Scholar]
- Modler, A.J.; Gast, K.; Lutsch, G.; Damaschun, G. Assembly of amyloid protofibrils via critical oligomers—A novel pathway of amyloid formation. J. Mol. Biol 2003, 325, 135–148. [Google Scholar]
- Gosal, W.S; Morten, L.J.; Hewitt, E.W.; Smith, D.A.; Thomson, N.H.; Radford, S.E. Competing pathways determine fibril morphology in the self-assembly of beta2-microglobulin into amyloid. J. Mol. Biol 2005, 351, 850–864. [Google Scholar]
- Smith, A.M.; Jahn, T.R.; Ashcroft, A.E.; Radford, S.E. Direct observation of oligomeric species formed in the early stages of amyloid fibril formation using electrospray ionisation mass spectrometry. J. Mol. Biol 2006, 364, 9–19. [Google Scholar]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Volles, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid beta-protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar]
- Baskakov, I.V.; Legname, G.; Baldwin, M.A.; Prusiner, S.B.; Cohen, F.E. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem 2002, 277, 21140–21148. [Google Scholar]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem 2007, 282, 10311–10324. [Google Scholar]
- McParland, V.J.; Kalverda, A.P.; Homans, S.W.; Radford, S.E. Structural properties of an amyloid precursor of beta(2)-microglobulin. Nat. Struct. Biol 2002, 9, 326–331. [Google Scholar]
- Lomas, D.A.; Carrell, R.W. Serpinopathies and the conformational dementias. Nat. Rev. Genet 2002, 3, 759–768. [Google Scholar]
- Serag, A.A.; Altenbach, C.; Gingery, M.; Hubbell, W.L.; Yeates, T.O. Arrangement of subunits and ordering of beta-strands in an amyloid sheet. Nat. Struct. Biol 2002, 9, 734–739. [Google Scholar]
- Guo, Z.; Eisenberg, D. Runaway domain swapping in amyloid-like fibrils of T7 endonuclease I. Proc. Natl. Acad. Sci. USA 2006, 103, 8042–8047. [Google Scholar]
- Rousseau, F.; Wilkinson, H.; Villanueva, J.; Serrano, L.; Schymkowitz, J.W.; Itzhaki, L.S. Domain swapping in p13suc1 results in formation of native-like, cytotoxic aggregates. J. Mol. Biol 2006, 363, 496–505. [Google Scholar]
- Plakoutsi, G.; Bemporad, F.; Calamai, M.; Taddei, N.; Dobson, C.M.; Chiti, F. Evidence for a mechanism of amyloid formation involving molecular reorganisation within native-like precursor aggregates. J. Mol. Biol 2005, 351, 910–922. [Google Scholar]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci 2007, 32, 204–206. [Google Scholar]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001, 29, 15–32. [Google Scholar]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid—From bacteria to humans. Trends Biochem. Sci 2007, 32, 217–224. [Google Scholar]
- Watt, J.A.; Pike, C.J.; Walencewicz-Wasserman, A.J.; Cotman, C.W. Ultrastructural analysis of beta-amyloid-induced apoptosis in cultured hippocampal neurons. Brain Res 1994, 661, 147–156. [Google Scholar]
- Bucciantini, M.; Rigacci, S.; Berti, A.; Pieri, L.; Cecchi, C.; Nosi, D.; Formigli, L.; Chiti, F.; Stefani, M. Patterns of cell death triggered in two different cell lines by HypF-N pre-fibrillar aggregates. FASEB J 2005, 19, 437–439. [Google Scholar]
- Ross, C.A. Polyglutamine pathogenesis: Emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 2002, 35, 819–822. [Google Scholar]
- Lane, N.J.; Balbo, A.; Fukuyama, R.; Rapoport, S.I.; Galdzick, Z. The ultrastructural effects of beta-amyloid peptide on cultured PC12 cells: Changes in cytoplasmic and intramembranous features. J. Neurocytol 1998, 27, 707–718. [Google Scholar]
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci 2001, 21, 7551–7560. [Google Scholar]
- Velez-Pardo, C.; Arroyave, S.T.; Lopera, F.; Castano, A.D.; Jimenez Del Rio, M. Ultrastructure evidence of necrotic neural cell death in familial Alzheimer’s disease brains bearing presenilin-1 E280A mutation. J. Alzheimers Dis 2001, 3, 409–415. [Google Scholar]
- Eremenko, E.; Ben-Zvi, A.; Morozova-Roche, L.A.; Raveh, D. Aggregation of human S100A8 and S100A9 amyloidogenic proteins perturbs proteostasis in a yeast model. PLoS One 2013, 8, e58218. [Google Scholar]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol 2013, 12, 105–118. [Google Scholar]
- Balietti, M.; Giorgetti, B.; Casoli, T.; Solazzi, M.; Tamagnini, F.; Burattini, C.; Aicardi, G.; Fattoretti, P. Early selective vulnerability of synapses and synaptic mitochondria in the hippocampal CA1 region of the Tg2576 mouse model of Alzheimer’s disease. J. Alzheimers Dis 2013, 34, 887–896. [Google Scholar]
- Caldeira, G.L.; Ferreira, I.L.; Rego, A.C. Impaired transcription in Alzheimer’s disease: Key role in mitochondrial dysfunction and oxidative stress. J. Alzheimers Dis 2013, 34, 115–131. [Google Scholar]
- Butterfield, A.D.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzeimer’s disease brain: Central role for amyloid β-peptide. Trends Mol. Med 2001, 7, 548–554. [Google Scholar]
- Sciacca, M.F.; Milardi, D.; Messina, G.M.; Marletta, G.; Brender, J.R.; Ramamoorthy, A.; La Rosa, C. Cations as switches of amyloid-mediated membrane disruption mechanisms: Calcium and IAPP. Biophys. J 2013, 104, 173–184. [Google Scholar]
- Villalobos, C.; Caballero, E.; Sanz-Blasco, S.; Núñez, L. Study of neurotoxic intracellular calcium signalling triggered by amyloids. Methods Mol. Biol 2012, 849, 289–302. [Google Scholar]
- Kourie, J.I. Mechanisms of amyloid β protein-induced modification in ion transport systems: Implications for neurodegenerative diseases. Cell. Mol. Neurobiol 2001, 21, 173–213. [Google Scholar]
- Sochocka, M.; Koutsouraki, E.S.; Gąsiorowski, K.; Leszek, J. Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer’s disease: New approach to therapy. CNS Neurol. Disord. Drug Targets 2013, in press. [Google Scholar]
- Milhavet, O.; Lehmann, S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res. Rev 2002, 38, 328–339. [Google Scholar]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-peptide 1–42-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal 2013, in press. [Google Scholar]
- Zhang, L.; Xing, G.Q.; Barker, J.L.; Chang, Y.; Maric, D.; Ma, W.; Li, B.-S.; Rubinow, D.R. α-Lipoic acid protects rat cortical neurons against cell death induced by amyloid and hydrogen peroxide through the Akt signalling pathway. Neurosci. Lett 2001, 312, 125–128. [Google Scholar]
- Yang, S.G.; Wang, W.Y.; Ling, T.J.; Feng, Y.; Du, X.T.; Zhang, X.; Sun, X.X.; Zhao, M.; Xue, D.; Yang, Y.; et al. α-Tocopherol quinone inhibits β-amyloid aggregation and cytotoxicity, disaggregates preformed fibrils and decreases the production of reactive oxygen species, NO and inflammatory cytokines. Neurochem. Int 2010, 57, 914–922. [Google Scholar]
- Zampagni, M.; Wright, D.; Cascella, R.; D’Adamio, G.; Casamenti, F.; Evangelisti, E.; Cardona, F.; Goti, A.; Nacmias, B.; Sorbi, S.; et al. Novel S-acyl glutathione derivatives prevent amyloid oxidative stress and cholinergic dysfunction in Alzheimer disease models. Free Radic. Biol. Med 2012, 52, 1362–1371. [Google Scholar]
- Choi, D.Y.; Lee, Y.J.; Hong, J.T.; Lee, H.J. Antioxidant properties of natural polyphenols and their therapeutic potentials for Alzheimer’s disease. Brain Res. Bull 2012, 87, 144–153. [Google Scholar]
- Rogers, I.; Kerr, F.; Martinez, P.; Hardy, J.; Lovestone, S.; Partridge, L. Ageing increases vulnerability to Aβ42 toxicity in Drosophila. PLoS One 2012, 7, e40569. [Google Scholar]
- Keller, J.N.; Huang, F.F.; Markesbery, W.R. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 2002, 98, 149–156. [Google Scholar]
- Fratta, P.; Engel, W.K.; McFerrin, J.; Davies, K.J.; Lin, S.W.; Askanas, V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am. J. Pathol 2005, 167, 517–526. [Google Scholar]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.-P.; Rubinsztein, D.C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by Huntingtin. Hum. Mol. Genet 2002, 11, 1137–1151. [Google Scholar]
- Wyttenbach, A.; Swartz, J.; Kita, H.; Thykjaer, T.; Carmichael, J.; Bradley, J.; Brown, R.; Maxwell, M.; Schapira, A.; Orntoft, T.F.; et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington’s disease. Hum. Mol. Genet 2001, 10, 1829–1845. [Google Scholar]
- Brunelle, P.; Rauk, A. The radical model of Alzheimer’s disease: Specific recognition of Gly29 and Gly33 by Met35 in a beta-sheet model of Abeta: An ONIOM study. J. Alzheimers Dis 2002, 4, 283–289. [Google Scholar]
- Orth, M.; Shapira, A.H.V. Mitochondrial involvement in Parkinson’s disease. Neurochem. Int 2002, 40, 533–541. [Google Scholar]
- Turnbull, S.; Tabner, B.J.; El-Agnaf, O.M.A.; Moore, S.; Davies, Y.; Allsop, D. α-Synuclein implicated in Parkinson’s disease catalyzes the formation of hydrogen peroxide in vitro. Free Rad. Biol. Med 2001, 30, 1163–1170. [Google Scholar]
- Tabner, B.J.; Turnbull, S.; El-Agnaf, O.M.A.; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Rad. Biol. Med 2002, 32, 1076–1083. [Google Scholar]
- Pappolla, M.A.; Omar, R.A.; Chyan, Y.-J.; Ghiso, J.; Hsiao, K.; Bozner, P.; Perry, G.; Smith, M.A.; Cruz-Sanchez, F. Induction of NADPH cytochrome P450 reductase by the Alzheimer β-protein. Amyloid as a “foreign body”. J. Neurochem 2001, 78, 121–128. [Google Scholar]
- Qin, L.; Liu, Y.; Cooper, C.; Liu, B.; Wilson, B.; Hong, J.-S. Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J. Neurochem 2002, 83, 973–983. [Google Scholar]
- Mattson, M.P. Impairment of membrane transport and signal transduction systems by amyloidogenic proteins. Methods Enzymol 1999, 309, 733–768. [Google Scholar]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Rego, A.C.; Oliveira, C. Effect of amyloid beta-peptide on permeability transition pore: A comparative study. J. Neurosci. Res 2002, 15, 257–267. [Google Scholar]
- Du, H.; Yan, S.S. Mitochondrial permeability transition pore in Alzheimer's disease: Cyclophilin D and amyloid beta. Biochim. Biophys. Acta 2010, 1802, 198–204. [Google Scholar]
- Squier, T.C. Oxidative stress and protein aggregation during biological aging. Exp. Gerontol 2001, 36, 1539–1550. [Google Scholar]
- Kawahara, M.; Kuroda, Y.; Arispe, N.; Rojas, E. Alzheimer’s β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevation by a common mechanism in a hypopthalamic GnRH neuronal cell line. J. Biol. Chem 2000, 275, 14077–14083. [Google Scholar]
- Sirangelo, I.; Malmo, C.; Iannuzzi, C.; Mezzogiorno, A.; Bianco, M.R.; Papa, M.; Irace, G. Fibrillogenesis and cytotoxic activity of the amyloid-forming apomyoglobin mutant W7FW14F. J. Biol. Chem 2004, 279, 13183–13189. [Google Scholar]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Pre-fibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem 2004, 279, 31374–31382. [Google Scholar]
- Cecchi, C.; Baglioni, S.; Fiorillo, C.; Pensalfini, A.; Liguri, G.; Nosi, D.; Rigacci, S.; Bucciantini, M.; Stefani, M. Insights into the molecular basis of the differing susceptibility of varying cell types to the toxicity of amyloid aggregates. J. Cell Sci 2005, 118, 3459–3470. [Google Scholar]
- Evangelisti, E.; Cecchi, C.; Cascella, R.; Sgromo, C.; Liguri, G.; Dobson, C.M.; Chiti, F.; Stefani, M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci 2012, 125, 2416–2427. [Google Scholar]
- Shadek, G.M.; Kummer, M.P.; Lu, D.C.; Galvan, V.; Bredesen, D.E.; Koo, E.H. Aβ induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597–624). FASEB J 2006, 20, 1254–1266. [Google Scholar]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Tumor necrosis factor death receptor signaling cascade Is required for amyloid-β protein-induced neuron death. J. Neurosci 2004, 24, 1760–1771. [Google Scholar]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhan, M.; Nagashima, J.; Morser, A.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar]
- Sasaki, N.; Takeuchi, M.; Chowei, H.; Kikuchi, S.; Hayashi, N.; Nakano, H.; Ikeda, S.; Yamagishi, T.; Kitamoto, T.; Saito, T.; et al. Advanced glycation end products (AGE) and their receptor (RAGE) in the brain of patients with Creutzfeldt-Jakob disease with prion plaques. Neurosci. Lett 2002, 326, 117–120. [Google Scholar]
- Monteiro, F.A.; Cardoso, I.; Mendes Sousa, M.; Saraiva, M.J. In vitro inhibition of transthyretin aggregate-induced cytotoxicity by full and peptide derived forms of the soluble receptor for advanced glycation end products (RAGE). FEBS Lett 2006, 580, 3451–3456. [Google Scholar]
- Deane, R.J. Is RAGE still a therapeutic target for Alzheimer’s disease? Future Med. Chem 2012, 4, 915–925. [Google Scholar]
- Geroldi, D.; Falcone, C.; Emanuele, E. Soluble receptor for advanced glycation end products: From disease marker to potential therapeutic target. Curr. Med. Chem 2006, 13, 1971–1978. [Google Scholar]
- Hou, X.; Parkington, H.C.; Coleman, H.A.; Mechler, A.; Martin, L.L.; Aguilar, M.-I.; Small, D.H. Transthyretin oligomers induce calcium influx via voltage-gated calcium channels. J. Neurochem 2007, 100, 446–457. [Google Scholar]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem 2007, 282, 11590–11601. [Google Scholar]
- Pellistri, F.; Bucciantini, M.; Relini, A.; Gliozzi, A.; Robello, M.; Stefani, M. Generic interaction of pre-fibrillar amyloid aggregates with NMDA and AMPA receptors results in free Ca2+ increase in primary neuronal cells. J. Biol. Chem 2008, 283, 29950–29960. [Google Scholar]
- Kranenburg, O.; Bouma, B.; Kroon-Batenburg, L.M.J.; Reijerkerk, A.; Wu, Y.-P.; Voest, E.E.; Gebbink, M.F.B.G. Tissue-type plasminogen activator is a multiligand cross-beta structure receptor. Curr. Biol 2002, 12, 1833–1839. [Google Scholar]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J. Neurosci 2004, 24, 10191–10200. [Google Scholar]
- Jayakumar, R.; Jayaraman, M.; Koteeswarl, D.; Gomath, K. Cytotoxic and membrane perturbation effects of a novel amyloid forming model, peptide poly(leucine-glutamic acid). J. Biochem 2004, 136, 457–462. [Google Scholar]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanisms of pathogenesis. Science 2003, 300, 486–489. [Google Scholar]
- Campioni, S.; Mannini, B.; Pensalfini, A.; Zampagni, M.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. Aberrant protein oligomer structures are causatively linked to cellular dysfunction. Nat. Chem. Biol 2010, 6, 140–147. [Google Scholar]
- Jarmuła, A.; Stępkowski, D. The β-sheet breakers and π-stacking. J. Pept. Sci 2013. [Google Scholar] [CrossRef]
- Luo, Y.; Vali, S.; Sun, S.; Chen, X.; Liang, X.; Drozhzhina, T.; Popugaeva, E.; Bezprozvanny, I. Aβ42-binding peptoids as amyloid aggregation inhibitors and detection ligands. ACS Chem. Neurosci 2013, in press. [Google Scholar]
- Parthsarathy, V.; McClean, P.L.; Hölscher, C.; Taylor, M.; Tinker, C.; Jones, G.; Kolosov, O.; Salvati, E.; Gregori, M.; Masserini, M.; et al. A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. PLoS One 2013, 8, e54769. [Google Scholar]
- Griffin, M.D.; Yeung, L.; Hung, A.; Todorova, N.; Mok, Y.F.; Karas, J.A.; Gooley, P.R.; Yarovsky, I.; Howlett, G.J. A cyclic peptide inhibitor of apoC.II peptide fibril formation: Mechanistic indight from NMR and molecular dynamics analysis. J. Mol. Biol 2012, 416, 642–655. [Google Scholar]
- Andreotti, G.; Vitale, R.M.; Avidan-Shpalter, C.; Amodeo, P.; Gazit, E.; Motta, A. Converting the highly amyloidogenic human calcitonin into a powerful fibril inhibitor by three-dimensional structure homology with a non-amyloidogenic analogue. J. Biol. Chem 2011, 286, 2707–2718. [Google Scholar]
- Soto, P.; Griffin, M.A.; Shea, J.E. New insights into the mechanism of Alzheimer amyloid-beta fibrillogenesis inhibition by N-methylated peptides. Biophys. J 2007, 93, 3015–3025. [Google Scholar]
- Costa, R.; Speretta, E.; Crowther, D.C.; Cardoso, I. Testing the therapeutic potential of doxycycline in a Drosophila melanogaster model of Alzheimer disease. J. Biol. Chem 2011, 286, 41647–41655. [Google Scholar]
- Ward, J.E.; Ren, R.; Toraldo, G.; Soohoo, P.; Guan, J.; O’Hara, C.; Jasuja, R.; Trinkaus-Randall, V.; Liao, R.; Connors, L.H.; et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood 2011, 118, 6610–6617. [Google Scholar]
- Giorgetti, S.; Raimondi, S.; Pagano, K.; Relini, A.; Bucciantini, M.; Corazza, A.; Fogolari, F.; Codutti, L.; Salmona, M.; Mangione, P.; et al. Effect of tetracyclines on the dynamics of formation and destructuration of beta2-microglobulin amyloid fibrils. J. Biol. Chem 2011, 286, 2121–2131. [Google Scholar]
- Aitken, J.F.; Loomes, K.M.; Scott, D.W.; Reddy, S.; Phillips, A.R.; Prijic, G.; Fernando, C.; Zhang, S.; Broadhurst, R.; L’Huillier, P.; et al. Tetracycline treatment retards the onset and slows the progression of diabetes in human amylin/islet amyloid polypeptide transgenic mice. Diabetes 2010, 59, 161–171. [Google Scholar]
- Lieu, V.H.; Wu, J.W.; Wang, S.S.; Wu, C.H. Inhibition of amyloid fibrillization of hen egg-white lysozymes by rifampicin and p-benzoquinone. Biotechnol. Prog 2007, 23, 698–706. [Google Scholar]
- Sant’Anna, R.O.; Braga, C.A.; Polikarpov, I.; Ventura, S.; Lima, L.M.T.R.; Foguel, D. Inhibition of human transthyretin aggregation by non-steroidal anti-inflammatory compounds: A structural and thermodynamic analysis. Int. J. Mol. Sci 2013, 14, 5284–5311. [Google Scholar]
- Legleiter, J.; Czilli, D.L.; Gitter, B.; DeMattos, R.B.; Holtzman, D.M.; Kowalewski, T. Effect of different anti-Abeta antibodies on Abeta fibrillogenesis as assessed by atomic force microscopy. J. Mol. Biol 2004, 335, 997–1006. [Google Scholar]
- Abe, M.; Abe, Y.; Ohkuri, T.; Mishima, T.; Monji, A.; Kanba, S.; Ueda, T. Mechanism for retardation of amyloid fibril formation by sugars in Vλ6 protein. Protein Sci 2013, 22, 467–474. [Google Scholar]
- Sirangelo, I.; Irace, G. Inhibition of aggregate formation as therapeutic target in protein misfolding diseases: Effect of tetracycline and trehalose. Expert Opin. Ther. Targets 2010, 14, 1311–1321. [Google Scholar]
- Huong, V.T.; Shimanouchi, T.; Shimauchi, N.; Yagi, H.; Umakoshi, H.; Goto, Y.; Kuboi, R. Catechol derivatives inhibit the fibril formation of amyloid-beta peptides. J. Biosci. Bioeng 2010, 109, 629–634. [Google Scholar]
- Mina, E.W.; Lasagna-Reeves, C.; Glabe, C.G.; Kayed, R. Poloxamer 188 copolymer membrane sealant rescues toxicity of amyloid oligomers in vitro. J. Mol. Biol 2009, 391, 577–585. [Google Scholar]
- Liu, F.F.; Ji, L.; Dong, X.Y.; Sun, Y. Molecular insight into the inhibition effect of trehalose on the nucleation and elongation of amyloid beta-peptide oligomers. J. Phys. Chem. B 2009, 113, 11320–11329. [Google Scholar]
- Morshedi, D.; Rezaei-Ghaleh, N.; Ebrahim-Habibi, A.; Ahmadian, S.; Nemat-Gorgani, M. Inhibition of amyloid fibrillation of lysozyme by indole derivatives-possible mechanism of action. FEBS J 2007, 274, 6415–6425. [Google Scholar]
- Yang, F., Jr; Zhang, M.; Zhou, B.R.; Chen, J.; Liang, Y. Oleic acid inhibits amyloid formation of the intermediate of alpha-lactalbumin at moderately acidic pH. J. Mol. Biol 2006, 362, 821–834. [Google Scholar]
- Pratim Bose, P.; Chatterjee, U.; Xie, L.; Johansson, J.; Göthelid, E.; Arvidsson, P.I. Effects of Congo red on Aβ(1–40) fibril formation process and morphology. ACS Chem. Neurosci 2010, 1, 315–324. [Google Scholar]
- Necula, M.; Breydo, L.; Milton, S.; Kayed, R.; van der Veer, W.E.; Tone, P.; Glabe, C.G. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry 2007, 46, 8850–8860. [Google Scholar]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.S.; Klang, I.M.; Lithgrow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar]
- Yao, J.; Ho, D.; Calingasan, N.Y.; Pipalia, N.H.; Lin, M.T.; Beal, M.F. Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. J. Exp. Med 2012, 209, 2501–2513. [Google Scholar]
- Cabaleiro-Lago, C.; Szczepankiewicz, O.; Linse, S. The effect of nanoparticles on amyloid aggregation depends on the protein stability and intrinsic aggregation rate. Langmuir 2012, 28, 1852–1857. [Google Scholar]
- Cabaleiro-Lago, C.; Lynch, I.; Dawson, K.A.; Linse, S. Inhibition of IAPP and IAPP(20–29) fibrillation by polymeric nanoparticles. Langmuir 2010, 26, 3453–3461. [Google Scholar]
- Pai, A.S.; Rubinstein, I.; Onyüksel, H. PEGylated phospholipid nanomicelles interact with beta-amyloid(1–42) and mitigate its beta-sheet formation, aggregation and neurotoxicity in vitro. Peptides 2006, 27, 2858–2866. [Google Scholar]
- Wang, S.S.; Chen, Y.T.; Chou, S.W. Inhibition of amyloid fibril formation of beta-amyloid peptides via the amphiphilic surfactants. Biochim. Biophys. Acta 2005, 1741, 307–313. [Google Scholar]
- Feng, B.Y.; Toyama, B.H.; Wille, H.; Colby, D.W.; Collins, S.R.; May, B.C.; Prusiner, S.B.; Weissman, J.; Shoichet, B.K. Small-molecule aggregates inhibit amyloid polymerization. Nat. Chem. Biol 2008, 4, 197–199. [Google Scholar]
- Török, B.; Sood, A.; Bag, S.; Kulkarni, A.; Borkin, D.; Lawler, E.; Dasgupta, S.; Landge, S.; Abid, M.; Zhou, W.; et al. Structure-activity relationships of organofluorine inhibitors of β-amyloid self-assembly. ChemMedChem 2012, 7, 910–919. [Google Scholar]
- Kolstoe, S.E.; Mangione, P.P.; Bellotti, V.; Taylor, G.W.; Tennent, G.A.; Deroo, S.; Morrison, A.J.; Cobb, A.J.; Coyne, A.; McCammon, M.G.; et al. Trapping of palindromic ligands within native transthyretin prevents amyloid formation. Proc. Natl. Acad. Sci. USA 2010, 107, 20483–20488. [Google Scholar]
- Castaño, A.; Helmke, S.; Alvarez, J.; Delisle, S.; Maurer, M.S. Diflunisal for ATTR cardiac amyloidosis. Congest. Heart Fail 2012, 18, 315–319. [Google Scholar]
- Bulawa, C.E.; Connelly, S.; Devit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington Cruz, M.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar]
- Berk, J.L.; Suhr, O.B.; Sekijima, Y.; Yamashita, T.; Heneghan, M.; Zeldenrust, S.R.; Ando, Y.; Ikeda, S.; Gorevic, P.; Merlini, G.; et al. Familial Amyloidosis Consortium. The Diflunisal Trial: Study accrual and drug tolerance. Amyloid 2012, 19, 37–38. [Google Scholar]
- Almeida, M.R.; Saraiva, M.J. Clearance of extracellular misfolded proteins in systemic amyloidosis: Experience with transthyretin. FEBS Lett 2012, 586, 2891–2896. [Google Scholar]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012, 19, 34–36. [Google Scholar]
- Masuda, M.; Hasegawa, M.; Nonaka, T.; Oikawa, T.; Yonetani, M.; Yamaguchi, Y.; Kato, K.; Hisanaga, S.; Goedert, M. Inhibition of alpha-synuclein fibril assembly by small molecules: Analysis using epitope-specific antibodies. FEBS Lett 2009, 583, 787–791. [Google Scholar]
- Demattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; Delong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar]
- Panza, F.; Frisardi, V.; Imbimbo, B.P.; Seripa, D.; Solfrizzi, V.; Pilotto, A. Monoclonal antibodies against β-amyloid (Aβ) for the treatment of Alzheimer’s disease: The Aβ target at a crossroads. Expert Opin. Biol. Ther 2011, 11, 679–686. [Google Scholar]
- Liu, Y.H.; Giunta, B.; Zhou, H.D.; Tan, J.; Wang, Y.J. Immunotherapy for Alzheimer disease: The challenge of adverse effects. Nat. Rev. Neurol 2012, 8, 465–469. [Google Scholar]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem 2002, 45, 1712–1722. [Google Scholar]
- McGovern, S.L.; Helfand, B.T.; Feng, B.; Shoichet, B.K. A specific mechanism of nonspecific inhibition. J. Med. Chem 2003, 46, 4265–4272. [Google Scholar]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J. Biol. Chem 2004, 279, 26846–26857. [Google Scholar]
- Blanchard, B.J.; Chen, A.; Rozeboom, L.M.; Stafford, K.A.; Weigele, P.; Ingram, V.M. Efficient reversal of Alzheimer’s disease fibril formation and elimination of neurotoxicity by a small molecule. Proc. Natl. Acad. Sci. USA 2004, 101, 14326–14332. [Google Scholar]
- Ferreira, N.; Santos, S.A.O.; Domingues, M.R.M.; Saraiva, M.J.; Almeida, M.R. Dietary curcumin counteracts extracellular transthyretin deposition: Insights on the mechanism of amyloid inhibition. Biochim. Biophys. Acta 2013, 1832, 39–45. [Google Scholar]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidoses: “In vivo” evidence from FAP mice models. PLoS One 2012, 7, e29933. [Google Scholar]
- Bieschke, J.; Hebst, M.; Wiglenda, T.; Friedrich, R.P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez del Amo, J.M.; Grüning, B.A.; Wang, Q.; et al. Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol 2011, 8, 93–101. [Google Scholar]
- Ladiwala, A.R.A.; Lin, J.C.; Bale, S.S.; Marcelino-Cruz, A.M.; Bhattacharya, M.; Dordick, J.S.; Tessier, P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem 2010, 285, 24228–24237. [Google Scholar]
- Ladiwala, A.R.A.; Dordick, J.S.; Tessier, P.M. Aromatic small molecules remodel toxic soluble oligomers of amyloid β through three independent pathways. J. Biol. Chem 2011, 286, 3209–3218. [Google Scholar]
- Ladiwala, A.R.A.; Mora-Pale, M.M.; Lin, J.C.; Bale, S.S.; Fishman, Z.S.; Dordick, J.S.; Tessier, P.M. Polyphenolic glycosides and aglycones utilize opposing pathways to selectively remodel and inactivate toxic oligomers of amyloid β. Chembiochem 2011, 12, 1749–1758. [Google Scholar]
- Queen, B.L.; Tollefsbol, T.O. Polyphenols and aging. Curr. Aging Sci 2010, 3, 34–42. [Google Scholar]
- Scarmeas, N.Y.; Stern, M.X.; Tang, R.; Mayeux, J.A.; Luchsinger, J.A. Mediterranean diet and risk for Alzheimer’s disease. Ann. Neurol 2006, 59, 912–921. [Google Scholar]
- Feart, C.; Samieri, C.; Barberger-Gateau, P. Mediterranean diet and cognitive function in older adults. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 14–18. [Google Scholar]
- Tangney, C.C.; Kwasny, M.J.; Li, H.; Wilson, R.S.; Evans, D.A.; Morris, M.C. Adherence to a Mediterranean-type dietary pattern and cognitive decline in a community population. Am. J. Clin. Nutr 2011, 93, 601–607. [Google Scholar]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci 2008, 65, 1631–1652. [Google Scholar]
- Ganguli, M.; Chandra, V.; Kamboh, M.I.; Johnston, J.M.; Dodge, H.H.; Thelma, B.K.; Juyal, R.C.; Pandav, R.; Belle, S.H.; DeKosky, S.T. Apolipoprotein E polymorphism and Alzheimer disease: The Indo-US Cross-National Dementia Study. Arch. Neurol 2000, 57, 824–830. [Google Scholar]
- Ng, T.P.; Chiam, P.C.; Lee, T.; Chua, H.C.; Lim, L.; Kua, E.H. Curry consumption and cognitive function in the elderly. Am. J. Epidemiol 2006, 164, 898–906. [Google Scholar]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.T.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem 2005, 280, 5892–5901. [Google Scholar]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci 2001, 21, 8370–8377. [Google Scholar]
- Hudson, S.A.; Ecroyd, H.; Kee, T.W.; Carver, J.A. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J 2009, 276, 5960–5972. [Google Scholar]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J. Neurosci. Res 2004, 75, 742–750. [Google Scholar]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Natural polyphenols inhibit different steps of the process of transthyretin (TTR) amyloid fibril formation. FEBS Lett 2011, 585, 2424–2430. [Google Scholar]
- Pullakhandam, R.; Srinivas, P.N.; Nair, M.K.; Reddy, G.B. Binding and stabilization of transthyretin by curcumin. Arch. Biochem. Biophys 2009, 485, 115–119. [Google Scholar]
- Caesar, I.; Jonson, M.; Nilsson, K.P.R.; Thor, S.; Hammarström, P. Curcumin promotes A-beta fibrillation and reduces neurotoxicity in transgenic drosophila. PLoS One 2012, 7, e31424. [Google Scholar]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am. J. Pathol 2009, 175, 2557–2565. [Google Scholar]
- Wang, S.S.-S.; Liu, K.-N.; Lee, W.-H. Effect of curcumin on the amyloid fibrillogenesis of hen egg-white lysozyme. Biophys. Chem 2009, 144, 78–87. [Google Scholar]
- Liu, K.-N.; Lai, C.-M.; Lee, Y.-T.; Wang, S.-N.; Chen, R.P.-Y.; Jan, J.-S.; Liu, H.-S.; Wang, S.S.-S. Curcumin’s pre-incubation temperature affects its inhibitory potency toward amyloid fibrillation and fibril-induced cytotoxicity of lysozyme. Biochim. Biophys. Acta 2012, 1820, 1774–1786. [Google Scholar]
- Sparks, S.; Liu, G.; Robbins, K.J.; Lazo, N.D. Curcumin modulates the self-assembly of the islet amyloid polypeptide by disassembling α-helix. Biochem. Biophys. Res. Commun 2012, 422, 551–555. [Google Scholar]
- Daval, M.; Bedrood, S.; Gurlo, T.; Huang, C.J.; Costes, S.; Butler, P.C.; Langen, R. The effect of curcumin on human islet amyloid polypeptide misfolding and toxicity. Amyloid 2010, 17, 118–128. [Google Scholar]
- Pandey, N.; Strider, J.V.; Nolan, W.C.; Yan, S.X.; Galvin, J.E. Curcumin inhibits aggregation of α-synuclein. Acta Neuropathol 2008, 115, 479–489. [Google Scholar]
- Bratkovič, I.H.; Gašperšič, J.; Šmid, L.M.; Bresjanac, M.; Jerala, R. Curcumin binds to the α-helical intermediate and to the amyloid form of prion protein—A new mechanism for the inhibition of PrPSc accumulation. J. Neurochem 2008, 104, 1553–1564. [Google Scholar]
- Yanagisawa, D.; Taguchi, H.; Yamamoto, A.; Shirai, N.; Hirao, K.; Tooyama, I. Curcuminoid binds to amyloid-β1–42 oligomer and fibril. J. Alzheimers Dis 2011, 24, 33–42. [Google Scholar]
- Ahmad, B.; Lapidus, L.J. Curcumin prevents aggregation in α-Synuclein by increasing reconfiguration rate. J. Biol. Chem 2012, 287, 9193–9199. [Google Scholar]
- Zhao, L.N.; Chiu, S.-W.; Benoit, J.; Chew, L.Y.; Mu, Y. The effect of curcumin on the stability of Aβ dimers. J. Phys. Chem. B 2012, 116, 7428–7435. [Google Scholar]
- Balasubramanian, K. Molecular orbital basis for yellow curry spice curcumin’s prevention of Alzheimer’s disease. J. Agric. Food Chem 2006, 54, 3512–3520. [Google Scholar]
- Masuda, Y.; Fukuchi, M.; Yatagawa, T.; Tada, M.; Takeda, T.; Irie, K.; Akagi, K.; Monobe, Y.; Imazawa, T.; Takegoshi, K. Solid-state NMR analysis of interaction sites of curcumin and 42-residue amyloid β-protein fibrils. Bioorg. Med. Chem 2011, 19, 5967–5974. [Google Scholar]
- Orlando, R.A.; Gonzales, A.M.; Royer, R.E.; Deck, L.M.; Vander Jagt, D.L. A chemical analog of curcumin as an improved inhibitor of amyloid abeta oligomerization. PLoS One 2012, 7, e31869. [Google Scholar]
- Reinke, A.A.; Gestwickiusing, J.E. Structure-activity relationships of amyloid beta-aggregation inhibitors based on curcumin: Influence of linker length and flexibility. Chem. Biol. Drug Des 2007, 70, 206–215. [Google Scholar]
- Ringman, J.M.; Frautschy, S.A.; Cole, G.M.; Masterman, D.L.; Cummings, J.L. A potential role of the curry spice curcumin in Alzheimer’s disease. Curr. Alzheimer Res 2005, 2, 131–136. [Google Scholar]
- Hamaguchi, T.; Ono, K.; Yamada, M. Curcumin and Alzheimer’s disease. CNS Neurosci. Ther. 2010, 16, 285–297. [Google Scholar]
- Monroy, A.; Lithgow, G.J.; Alavez, S. Curcumin and neurodegenerative diseases. IUBMB Inc 2013, 39, 122–132. [Google Scholar]
- Choi, Y.T.; Jung, C.H.; Lee, S.R.; Bae, J.H.; Baek, W.K.; Suh, M.H.; Park, J.; Park, C.W.; Suh, S.I. The green tea polyphenol (−)-epigallocatechin gallate attenuates beta-amyloid-induced neurotoxicity in cultured hippocampal neurons. Life Sci 2001, 70, 603–614. [Google Scholar]
- Rezai-Zadeh, K.; Shytle, D.; Sun, N.; Mori, T.; Hou, H.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D.; et al. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J. Neurosci 2005, 25, 8807–8814. [Google Scholar]
- Kuriyama, S.; Hozawa, A.; Ohmori, K.; Shimazu, T.; Matsui, T.; Ebihara, S.; Awata, S.; Nagatomi, R.; Arai, H.; Tsuji, I. Green tea consumption and cognitive function: A cross-sectional study from the Tsurugaya Project 1. Am. J. Clin. Nutr 2006, 83, 355–361. [Google Scholar]
- Reznichenko, L.; Amit, T.; Zheng, H.; Avramovich-Tirosh, Y.; Youdim, M.B.; Weinreb, O.; Mandel, S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (−)-epigallocatechin-3-gallate in cell cultures: Implications for iron chelation in Alzheimer’s disease. J. Neurochem 2006, 97, 527–536. [Google Scholar]
- Bastianetto, S.; Yao, Z.X.; Papadopoulos, V.; Quirion, R. Neuroprotective effects of green and black teas and their catechin gallate esters against beta-amyloid-induced toxicity. Eur. J. Neurosci 2006, 23, 55–64. [Google Scholar]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet 2006, 15, 2743–2751. [Google Scholar]
- Ferreira, N.; Cardoso, I.; Domingues, M.R.; Vitorino, R.; Bastos, M.; Bai, G.; Saraiva, M.J.; Almeida, M.R. Binding of epigallocatechin-3-gallate to transthyretin modulates its amyloidogenicity. FEBS Lett. 2009, 583, 3569–3576. [Google Scholar]
- Meng, F.; Abedini, A.; Plesner, A.; Verchere, C.B.; Raleigh, D.P. The flavanol (−)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry 2010, 49, 8127–8133. [Google Scholar]
- Hauber, I.; Hohenberg, H.; Holstermann, B.; Hunstein, W.; Hauber, J. The main green tea polyphenol epigallocatechin-3-gallate counteracts semen-mediated enhancement of HIV infection. Proc. Natl. Acad. Sci. USA 2009, 106, 9033–9038. [Google Scholar]
- He, J.; Xing, Y.-F.; Huang, B.; Zhang, Y.-Z.; Zeng, C.-M. Tea catechins induce the conversion of preformed lysozyme amyloid fibrils to amorphous aggregates. J. Agric. Food Chem 2009, 57, 11391–11396. [Google Scholar]
- Hudson, S.A.; Ecroyd, H.; Dehle, F.C.; Musgrave, I.F.; Carver, J.A. (−)-Epigallocatechin-3-Gallate (EGCG) maintains κ-casein in its pre-fibrillar state without redirecting its aggregation pathway. J. Mol. Biol 2009, 392, 689–700. [Google Scholar]
- Huang, R.; Vivekanandan, S.; Brender, J.R.; Abe, Y.; Naito, A.; Ramamoorthy, A. NMR characterization of monomeric and oligomeric conformations of human calcitonin and its interaction with EGCG. J. Mol. Biol 2012, 416, 108–120. [Google Scholar]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol 2008, 15, 558–566. [Google Scholar]
- Cao, P.; Raleigh, D.P. Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry 2012, 51, 2670–2683. [Google Scholar]
- Ghosh, S.; Pandey, N.K.; Dasgupta, S. (−)-Epicatechin gallate prevents alkali-salt mediated fibrillogenesis of hen egg white lysozyme. Int. J. Biol. Macromol 2012, 54, 90–98. [Google Scholar]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar]
- Lopez del Amo, J.M.; Fink, U.; Dasari, M.; Grelle, G.; Wanker, E.E.; Bieschke, J.; Reif, B. Structural properties of EGCG-induced, nontoxic Alzheimer’s disease Aβ oligomers. J. Mol. Biol 2012, 421, 517–524. [Google Scholar]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am Chem. Soc 2013, 135, 7503–7510. [Google Scholar]
- Wang, S.-H.; Liu, F.-F.; Dong, X.Y.; Sun, Y. Thermodynamic analysis of the molecular interactions between amyloid β-peptide 42 and (−)-epigallocatechin-3-gallate. J. Phys. Chem. B 2010, 114, 11576–11583. [Google Scholar]
- Wang, S.-H.; Dong, X.-Y.; Sun, Y. Thermodynamic analysis of the molecular interactions between amyloid β-protein fragments and (−)-epigallocatechin-3-gallate. J. Phys. Chem. B 2012, 116, 5803–5809. [Google Scholar]
- Kamihira-Ishijima, M.; Nakazawa, H.; Kira, A.; Naito, A.; Nakayama, T. Inhibitory mechanism of pancreatic amyloid fibril formation: Formation of the complex between tea catechins and the fragment of residues 22–27. Biochemistry 2012, 51, 10167–10174. [Google Scholar]
- Engel, M.F.M.; vandenAkker, C.C.; Schleeger, M.; Velikov, K.P.; Koenderink, G.H.; Bonn, M. The polyphenol EGCG inhibits amyloid formation less efficiently at phospholipid interfaces than in bulk solution. J. Am. Chem. Soc 2012, 134, 14781–14788. [Google Scholar]
- Uekusa, Y.; Kamihira-Lshijima, M.; Sugimoto, O.; Ishii, T.; Kumazawa, S.; Nakamura, K.; Tanji, K.; Naito, A.; Nakayama, T. Interaction of epipcatechin gallate with phospholipid membranes as revealed by solid-state NMR spectroscopy. Biochim. Biophys. Acta 2011, 1808, 1654–1660. [Google Scholar]
- Barry, J.; Fritz, M.; Brender, J.R.; Smith, P.E.S.; Lee, D.K.; Ramamoorthy, A. Determining the effects of lipophilic drugs on membrane structure by solid-state NMR spectroscopy: The case of the antioxidant curcumin. J. Am. Chem. Soc 2009, 131, 4490–4498. [Google Scholar]
- Popovych, N.; Brender, J.R.; Soong, R.; Vivekanandan, S.; Hartman, K.; Basrur, V.; Macdonald, P.M.; Ramamoorthy, A. Site specific interaction of the polyphenol EGCG with the SEVI amyloid precursor peptide PAP(248–286). J. Phys. Chem. B 2012, 116, 3650–3658. [Google Scholar]
- Miyata, M.; Sato, T.; Kugimiya, M.; Sho, M.; Nakamura, T.; Ikemizu, S.; Chirifu, M.; Mizuguchi, M.; Nabeshima, Y.; Suwa, Y.; et al. The crystal structure of the green tea polyphenol (−)-epigallocatechin gallate-transthyretin complex reveals a novel binding site distinct from the thyroxine binding site. Biochemistry 2010, 49, 6104–6114. [Google Scholar]
- Lee, M.J.; Maliakal, P.; Chen, L.; Meng, X.; Bondoc, F.Y.; Prabhu, S.; Lambert, G.; Mohr, S.; Yang, C.S. Pharmacokinetics of tea catechins after ingestion of green tea and (−)-epigallocatechin-3-gallate by humans: Formation of different metabolites and individual variability. Cancer Epidemiol. Biomark. Prev 2002, 11, 1025–1032. [Google Scholar]
- Mereles, D.; Hunstein, W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int. J. Mol. Sci 2011, 12, 5592–5603. [Google Scholar]
- Giunta, B.; Hou, H.; Zhu, Y.; Salemi, J.; Ruscin, A.; Shytle, R.D.; Tan, J. Fish oil enhances anti-amyloidogenic properties of green tea EGCG in Tg2576 mice. Neurosci. Lett 2010, 471, 134–138. [Google Scholar]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug. Discov 2006, 5, 493, e506.. [Google Scholar]
- Orgogozo, J.M.; Dartigues, J.F.; Lafont, S.; Letenneur, L.; Commenges, D.; Salamon, R.; Renaud, S.; Breteler, M.B. Wine consumption and dementia in the elderly: A prospective community study in the Bordeaux area. Rev. Neurol 1997, 153, 185–192. [Google Scholar]
- Vingtdeux, V.; Dreses-Werringloer, U.; Zhao, H.; Davies, P.; Marambaud, P. Therapeutic potential of resveratrol in Alzheimer’s disease. BMC Neurosci 2008, 9, S6. [Google Scholar]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.-L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int 2009, 54, 111–118. [Google Scholar]
- Lu, C.; Guo, Y.; Li, J.; Yao, M.; Liao, Q.; Xie, Z.; Li, X. Design, synthesis, and evaluation of resveratrol derivatives as Aβ1–42 aggregation inhibitors, antioxidants, and neuroprotective agents. Bioorg. Med. Chem. Lett 2012, 22, 7683–7687. [Google Scholar]
- Frozza, R.L.; Bernardi, A.; Hoppe, J.B.; Meneghetti, A.B.; Matté, A.; Battastini, A.M.O.; Pohlmann, A.R.; Guterres, S.S.; Salbego, C. Neuroprotective effects of resveratrol against Aβ administration in rats are improved by lipid-core nanocapsules. Mol. Neurobiol. 2003. [Google Scholar] [CrossRef]
- Amria, A.; Chaumeila, J.C.; Sfarb, S.; Charrueau, C. Administration of resveratrol: What formulation solutions to bioavailability limitations? J. Control Release 2012, 158, 182–193. [Google Scholar]
- Jang, M.H.; Piao, X.L.; Kim, H.Y.; Cho, E.J.; Baek, S.H.; Kwon, S.W.; Park, H.L. Resveratrol oligomers from vitis amurensis attenuate β-amyloid-induced oxidative stress in PC12 cells. Biol. Pharm. Bull 2007, 30, 1130–1134. [Google Scholar]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-β peptides. J. Biol. Chem 2005, 280, 37377–37382. [Google Scholar]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent β-amyloid toxicity through inhibiting NF-κB signaling. J. Biol. Chem 2005, 280, 40364–40374. [Google Scholar]
- Anekonda, T.S.; Reddy, P.H. Neuronal protection by sirtuins in Alzheimer’s disease. J. Neurochem 2006, 96, 305, e13.. [Google Scholar]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J. Biol. Chem 2010, 285, 9100–9113. [Google Scholar]
- Han, Y.S.; Bastianetto, S.; Dumont, Y.; Quirion, R. Specific plasma membrane binding sites for polyphenols, including resveratrol, in the rat brain. J. Pharmacol. Exp. Ther 2006, 318, 238–245. [Google Scholar]
- Mishra, R.; Sellin, D.; Radovan, D.; Gohlke, A.; Winter, R. Inhibiting islet amyloid polypeptide fibril formation by the red wine compound resveratrol. Chembiochem 2009, 10, 445–449. [Google Scholar]
- Evers, F.; Jeworrek, C.; Tiemeyer, S.; Weise, K.; Sellin, D.; Paulus, M.; Struth, B.; Tolan, M.; Winter, R. Elucidating the mechanism of lipid membrane-induced IAPP fibrillogenesis and its inhibition by the red wine compound resveratrol: A synchrotron X-ray reflectivity study. J. Am. Chem. Soc 2009, 131, 9516–9521. [Google Scholar]
- Sparr, E.; Engel, M.F.M.; Sakharov, D.V.; Sprong, M.; Jacobs, J.; de Kruijff, B.; Höppener, J.W.M.; Killian, J.A. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett 2004, 577, 117–120. [Google Scholar]
- Radovan, D.; Opitz, N.; Winter, R. Fluorescence microscopy studies on islet amyloid polypeptide fibrillation at heterogeneous and cellular membrane interfaces and its inhibition by resveratrol. FEBS Lett 2009, 583, 1439–1445. [Google Scholar]
- Wei, L.; Jiang, P.; Xu, W.; Li, H.; Zhang, H.; Yan, L.; Chan-Park, M.B.; Liu, X.-W.; Tang, K.; Mu, Y.; et al. The molecular basis of distinct aggregation pathways of islet amyloid polypeptide. J. Biol. Chem 2011, 286, 6291–6300. [Google Scholar]
- Feng, Y.; Wanga, X.-P.; Yang, S.-G.; Wang, Y.-J.; Zhang, X.; Du, X.-T.; Sun, X.-X.; Zhao, M.; Huang, L.; Liu, R.-T. Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 2009, 30, 986–995. [Google Scholar]
- Ge, J.-F.; Qiao, J.-P.; Qi, C.-C.; Wang, C.-W.; Zhou, J.-N. The binding of resveratrol to monomer and fibril amyloid beta. Neurochem. Int 2012, 61, 1192–1201. [Google Scholar]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol 2000, 7, 312–321. [Google Scholar]
- Gazova, Z.; Siposova, K.; Kurin, E.; Mučaji, P.; Nagy, M. Amyloid aggregation of lysozyme: The synergy study of red wine polyphenols. Proteins 2013. [Google Scholar] [CrossRef]
- Kim, H.; Park, B.-S.; Lee, K.-G.; Choi, C.Y.; Jang, S.S.; Kim, Y.-H.; Lee, S.-E. Effects of naturally occurring compounds on fibril formation and oxidative stress of β-amyloid. J. Agric. Food Chem 2005, 53, 8537–8541. [Google Scholar]
- Akaishi, T.; Morimoto, T.; Shibao, M.; Watanabe, S.; Sakai-Kato, K.; Utsunomiya-Tate, N.; Abe, K. Structural requirements for the flavonoid fisetin in inhibiting fibril formation of amyloid β protein. Neurosci. Lett 2008, 444, 280–285. [Google Scholar]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Multifunction of myricetin on Aβ: Neuroprotection via a conformational change of Aβ and reduction of Aβ Via the interference of secretases. J. Neurosci. Res 2008, 86, 368–377. [Google Scholar]
- Wang, J.B.; Wang, Y.M.; Zeng, C.M. Quercetin inhibits amyloid fibrillation of bovine insulin and destabilizes preformed fibrils. Biochem. Biophys. Res. Commun 2011, 415, 675–679. [Google Scholar]
- Ono, K.; Yamada, M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for α-synuclein fibrils in vitro. J. Neurochem 2006, 97, 105–115. [Google Scholar]
- Jagota, S.; Rajadas, J. Effect of phenolic compounds against Aβ aggregation and Aβ-induced toxicity in transgenic C. elegans. Neurochem. Res 2012, 37, 40–48. [Google Scholar]
- Noor, H.; Cao, P.; Raleigh, D.P. Morin hydrate inhibits amyloid formation by islet amyloid polypeptide and disaggregates amyloid fibers. Protein Sci 2012, 21, 373–382. [Google Scholar]
- Zelus, C.; Fox, A.; Calciano, A.; Faridian, B.S.; Nogaj, L.A.; Moffet, D.A. Myricetin Inhibits islet amyloid polypeptide (IAPP) aggregation and rescues living mammalian cells from IAPP toxicity. Open Biochem. J 2012, 6, 66–70. [Google Scholar]
- Hirohata, M.; Hasegawa, K.; Tsutsumi-Yasuhara, S.; Ohhashi, Y.; Ookoshi, T.; Ono, K.; Yamada, M.; Naiki, H. The anti-amyloidogenic effect is exerted against Alzheimer’s β-amyloid fibrils in vitro by preferential and reversible binding of flavonoids to the amyloid fibril structure. Biochemistry 2007, 46, 1888–1899. [Google Scholar]
- Bartolini, M.; Naldi, M.; Fiori, J.; Valle, F.; Biscarini, F.; Nicolau, D.V.; Andrisano, V. Kinetic characterization of amyloid-beta 1–42 aggregation with a multimethodological approach. Anal. Biochem 2011, 414, 215–225. [Google Scholar]
- Fiori, J.; Naldi, M.; Bartolini, M.; Andrisano, V. Disclosure of a fundamental clue for the elucidation of the myricetin mechanism of action as amyloid aggregation inhibitor by mass spectrometry. Electrophoresis 2012, 33, 3380–3386. [Google Scholar]
- Ono, K.; Li, L.; Takamura, Y.; Yoshiike, Y.; Zhu, L.; Han, F.; Mao, X.; Ikeda, T.; Takasaki, J.; Nishijo, H.; et al. Phenolic compounds prevent amyloid β-protein oligomerization and synaptic dysfunction by site-specific binding. J. Biol. Chem 2012, 287, 14631–14643. [Google Scholar]
- Berhanu, W.M.; Masunov, A.E. Natural polyphenols as inhibitors of amyloid aggregation. Molecular dynamics study of GNNQQNY heptapeptide decamer. Biophys. Chem 2010, 149, 12–21. [Google Scholar]
- Wang, M.; Jiji, R.D. Resolution of localized small molecule-Aβ interactions by deep-ultraviolet resonance Raman spectroscopy. Biophys. Chem 2011, 158, 96–103. [Google Scholar]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Flavonols and flavones as BACE-1 inhibitors: Structure-activity relationship in cell-free, cell-based and in silico studies reveal novel pharmacophore features. Biochi. Biophys. Acta 2008, 1780, 819–825. [Google Scholar]
- Ansari, M.A.; Abdul, H.M.; Joshi, G.; Opii, W.O.; Butterfield, D.A. Protective effect of quercetin in primary neurons against Aβ(1–42): Relevance to Alzheimer’s disease. J. Nutr. Biochem 2009, 20, 269–275. [Google Scholar]
- Shi, C.; Zhao, L.; Zhu, B.; Li, Q.; Yew, D.T.; Yao, Z.; Xu, J. Protective effects of Ginkgo biloba extract (EGb761) and its constituents quercetin and ginkgolide B against β-amyloid peptide-induced toxicity in SH-SY5Y cells. Chem. Biol. Interact 2009, 181, 115–123. [Google Scholar]
- Pocernich, C.B.; Lange, M.L.; Sultana, R.; Butterfield, D.A. Nutritional approaches to modulate oxidative stress in Alzheimer’s disease. Curr. Alzheimer Res 2011, 8, 452–469. [Google Scholar]
- Tay, W.M.; da Silva, G.F.Z.; Ming, L.-J. Metal binding of flavonoids and their distinct inhibition mechanisms toward the oxidation activity of Cu2+-β-amyloid: Not just serving as suicide antioxidants! Inorg. Chem 2013, 52, 679–690. [Google Scholar]
- DeToma, A.S.; Choi, J.-S.; Braymer, J.J.; Lim, M.H. Myricetin: A naturally occurring regulator of metal-induced amyloid-β aggregation and neurotoxicity. Chembiochem 2011, 12, 1198–1201. [Google Scholar]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Zhang, Z.F.; Shan, Q.; Zheng, Z.H.; Liu, C.M.; Wang, Y.J. Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J. Pathol 2010, 222, 199–212. [Google Scholar]
- Tchantchou, F.; Lacor, P.N.; Cao, Z.; Lao, L.; Hou, Y.; Cui, C.; Klein, W.L.; Luo, Y. Stimulation of neurogenesis and synaptogenesis by bilobalide and quercetin via common final pathway in hippocampal neurons. J. Alzheimers Dis 2009, 18, 787–798. [Google Scholar]
- Tedeschi, A.; D’Errico, G.; Lauro, M.R.; Sansone, F.; Di Marino, S.; D’Ursi, A.M.; Aquino, R.P. Effect of flavonoids on the Abeta(25–35)-phospholipid bilayers interaction. Eur. J. Med. Chem 2010, 45, 3998–4003. [Google Scholar]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Three distinct neuroprotective functions of myricetin against glutamate-induced neuronal cell death: Involvement of direct inhibition of caspase-3. J. Neurosci. Res 2008, 86, 1836–1845. [Google Scholar]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem 2005, 280, 7614–7623. [Google Scholar]
- Pitozzi, V.; Jacomelli, M.; Zaid, M.; Luceri, C.; Bigagli, E.; Lodovici, M.; Ghelardini, C.; Vivoli, E.; Norcini, M.; Gianfriddo, M.; et al. Effects of dietary extra-virgin olive oil on behaviour and brain biochemical parameters in ageing rats. Br. J. Nutr 2010, 11, 1674–1683. [Google Scholar]
- Farr, .SA.; Price, T.O.; Dominguez, L.J.; Motisi, A.; Saiano, F.; Niehoff, M.L.; Morley, J.E.; Banks, W.A.; Ercal, N.; Barbagallo, M. Extra virgin olive oil improves learning and memory in SAMP8 mice. J. Alzheimers Dis 2012, 28, 81–92. [Google Scholar]
- Berr, C.; Portet, F.; Carriere, I.; Akbaraly, T.N.; Feart, C.; Gourlet, V.; Combe, N.; Barberger-Gateau, P.; Ritchie, K. Olive oil and cognition: Results from the three-city study. Dement. Geriatr. Cogn. Disord 2009, 4, 357–364. [Google Scholar]
- Brenes, M.; García, A.; García, P.; Rios, J.J.; Garrido, A. Phenolic compounds in spanish olive oils. J. Agric. Food Chem 1999, 47, 3535–3540. [Google Scholar]
- Romani, A.; Mulinacci, N.; Pinelli, P.; Vincieri, F.F.; Cimato, A. Polyphenolic content in five Tuscany cultivars of Olea europaea L. J. Agric. Food Chem 1999, 47, 964–967. [Google Scholar]
- Franconi, F.; Coinu, R.; Carta, S.; Urgeghe, P.P.; Ieri, F.; Mulinacci, N.; Romani, A. Antioxidant effect of two virgin olive oils depends on the concentration and composition of minor polar compounds. J. Agric. Food Chem 2006, 54, 3121–3125. [Google Scholar]
- Tripoli, E.; Giammanco, M.; Tabacchi, G.; Di Majo, D.; Giammanco, S.; La Guardia, M. The phenolic compounds of olive oil: Structure, biological activity and beneficial effects on human health. Nutr. Res. Rev 2005, 18, 98–112. [Google Scholar]
- Cicerale, S.; Conlan, X.A.; Sinclair, A.J.; Keast, R.S.J. Chemistry and health of olive oil phenolics. Crit. Rev. Food Sci. Nutr 2009, 49, 218–236. [Google Scholar]
- Bazoti, F.N.; Berquist, J.; Markides, K.E.; Tsarbopoulos, A. Noncovalent interaction between amyloid-β-peptide (1–40) and oleuropein studied by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom 2006, 17, 568–575. [Google Scholar]
- Bazoti, F.N.; Berquist, J.; Markides, K.E.; Tsarbopoulos, A. Localization of the noncovalent binding site between amyloid-β-peptide and oleuropein using electrospray ionization FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom 2008, 19, 1078–1085. [Google Scholar]
- Kallberg, Y.; Gustafsson, M.; Persson, B.; Thyberg, J.; Johansson, J.J. Prediction of amyloid fibril-forming proteins. J. Biol. Chem 2001, 276, 12945–12950. [Google Scholar]
- Tjernberg, L.O.; Callaway, D.J.E.; Tjernberg, A.; Hahne, S.; Lilliehöök, K.; Terenius, L.; Thyberg, J.; Nordstedt, C. A molecular model of Alzheimer amyloid beta-peptide fibril formation. J. Biol. Chem 1999, 274, 12619–12625. [Google Scholar]
- Tjernberg, L.O.; Näslund, L.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C.J. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. Biol. Chem 1996, 271, 8545–8548. [Google Scholar]
- Benaki, D.; Stathopoulou, K.; Leondiadis, L.; Ferderigos, N.; Pelecanou, M.; Mikros, E. Detection of interactions of the β-amyloid peptide with small molecules employing transferred NOEs. J. Pept. Sci 2009, 15, 435–441. [Google Scholar]
- Galanakis, P.A.; Bazoti, F.N.; Bergquist, J.; Markides, K.; Spyroulias, G.A.; Tsarbopoulos, A. Study of the interaction between the amyloid beta peptide (1–40) and antioxidant compounds by nuclear magnetic resonance spectroscopy. Biopolymers 2011, 96, 316–327. [Google Scholar]
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Parri, M.; Nediani, C.; Cerbai, E.; Stefani, M.; Berti, A. Oleuropein aglycon prevents cytotoxic amyloid aggregation of human amylin. J. Nutr. Biochem 2010, 21, 726–735. [Google Scholar]
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Nichino, D.; Relini, A.; Berti, A.; Stefani, M. Aβ(1–42) aggregates into non-toxic amyloid assemblies in the presence of the natural polyphenol oleuropein aglycon. Curr. Alzheimer Res 2011, 8, 841–852. [Google Scholar]
- Diomede, L.; Rigacci, S.; Romeo, M.; Stefani, M.; Salmona, M. Oleuropein aglycone protects transgenic C. elegans strains expressing Aβ42 by reducing plaque load and motor deficit. PLoS One 2013, 8, e58893. [Google Scholar]
- Grossi, C.; Rigacci, S.; Ambrosini, S.; Ed Dami, T.; Luccarini, I.; Traini, C.; Failli, P.; Berti, A.; Casamenti, F.; Stefani, M. The polyphenol oleuropein aglycone protects TgCRND8 mice against Aβ plaque pathology. 2013. Submitted for publication. [Google Scholar]
- Daccache, A.; Lion, C.; Sibille, N.; Gerard, M.; Slomianny, C.; Lippens, G.; Cotelle, P. Oleuropein and derivatives from olives as Tau aggregation inhibitors. Neurochem. Int 2011, 58, 700–707. [Google Scholar]
- Kostomoiri, M.; Fragkouli, A.; Sagnou, M.; Skaltsounis, L.A.; Pelecanou, M.; Tsilibary, E.C.; Szinia, A.K. Oleuropein, an anti-oxidant polyphenol constituent of olive promotes α-secretase cleavage of the amyloid precursor protein (AβPP). Cell. Mol. Neurobiol 2013, 33, 147–154. [Google Scholar]
- Pitt, J.; Roth, W.; Lacor, P.; Smith, A.B., III; Blankenship, M.; Velasco, P.; De Felice, F.; Breslin, P.; Klein, W.L. Alzheimer’s-associated Aβ oligomers show altered structure, immunoreactivity and synaptotoxicity with low doses of oleocanthal. Toxicol. Appl. Pharmacol 2009, 240, 189–197. [Google Scholar]
- Li, W.; Sperry, J.B.; Crowe, A.; Trojanowski, J.Q.; Smith, A.B., III; Lee, V.M.-Y. Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau. J. Neurochem 2009, 110, 1339–1351. [Google Scholar]
- Monti, M.C.; Margarucci, L.; Riccio, R.; Casapullo, A. Modulation of Tau protein fibrillization by Oleocanthal. J. Nat. Prod 2012, 75, 1584–1588. [Google Scholar]
- Abuznait, A.H.; Qosa, H.; Busnena, B.A.; El Sayed, K.A.; Kaddoumi, A. Olive-oil-derived oleocanthal enhances β-amyloid clearance as a potential neuroprotective mechanism against Alzheimer’s disease: In vitro and in vivo studies. ACS Chem. Neurosci 2013. [Google Scholar] [CrossRef]
- Serra, A.; Rubio, L.; Borras, X.; Macia, A; Romero, M.P.; Motilva, M.J. Distribution of olive oil phenolic compounds in rat tissues after administration of a phenolic extract from olive cake. Mol. Nutr. Food Res 2012, 3, 486–496. [Google Scholar]
- Vissers, M.N.; Zock, P.L.; Roodenburg, A.J.C.; Leenen, R.; Katan, M.B. Olive oil phenols are absorbed in humans. J. Nutr 2002, 132, 409–417. [Google Scholar]
- Ono, K.; Hasegawa, K.; Yoshiike, Y.; Takashima, A.; Yamada, M.; Naiki, H. Nordihydroguaiaretic acid potently breaks down pre-formed Alzheimer’s beta-amyloid fibrils in vitro. J. Neurochem 2002, 81, 434–440. [Google Scholar]
- Moss, M.A.; Varvel, N.H.; Nichols, M.R.; Reed, D.K.; Rosenberry, T.L. Nordihydroguaiaretic acid does not disaggregate beta-amyloid(1–40) protofibrils but does inhibit growth arising from direct protofibril association. Mol. Pharmacol 2004, 66, 592–600. [Google Scholar]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer’s beta-amyloid fibrils in vitro. Biochim. Biophys. Acta 2004, 1690, 193–202. [Google Scholar]
- Feng, Y.; Yang, S.G.; Du, X.T.; Zhang, X.; Sun, X.X.; Zhao, M.; Sun, G.Y.; Liu, R.T. Ellagic acid promotes Abeta42 fibrillization and inhibits Abeta42-induced neurotoxicity. Biochem. Biophys. Res. Commun 2009, 390, 1250–1254. [Google Scholar]
- Gauci, A.J.; Caruana, M.; Giese, A.; Scerri, C.; Vassallo, N. Identification of polyphenolic compounds and black tea extract as potent inhibitors of lipid membrane destabilization by Aβ42 aggregates. J. Alzheimers Dis 2011, 27, 767–779. [Google Scholar]
- Zhao, L.; Wang, J.L.; Wang, Y.R.; Fa, X.Z. Apigenin attenuates copper-mediated β-amyloid neurotoxicity through antioxidation, mitochondrion protection and MAPK signal inactivation in an AD cell model. Brain Res 2013, 1492, 33–45. [Google Scholar]
- Bae, S.Y.; Kim, S.; Hwang, H.; Kim, H.K.; Yoon, H.C.; Kim, J.H.; Lee, S.; Kim, T.D. Amyloid formation and disaggregation of α-synuclein and its tandem repeat (α-TR). Biochem. Biophys. Res. Commun 2010, 400, 531–536. [Google Scholar]
- Lu, J.H.; Ardah, M.T.; Durairajan, S.S.; Liu, L.F.; Xie, L.X.; Fong, W.F.; Hasan, M.Y.; Huang, J.D.; El-Agnaf, O.M.; Li, M. Baicalein inhibits formation of α-synuclein oligomers within living cells and prevents Aβ peptide fibrillation and oligomerisation. Chembiochem 2011, 12, 615–624. [Google Scholar]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem 2003, 87, 172–181. [Google Scholar]
- Lemkul, J.A.; Bevan, D.R. Destabilizing Alzheimer’s Abeta(42) protofibrils with morin: Mechanistic insights from molecular dynamics simulations. Biochemistry 2010, 49, 3935–3946. [Google Scholar]
- Ushikubo, H.; Watanabe, S.; Tanimoto, Y.; Abe, K.; Hiza, A.; Ogawa, T.; Asakawa, T.; Kan, T.; Akaishi, T. 3,3′,4′,5,5′-Pentahydroxyflavone is a potent inhibitor of amyloid β fibril formation. Neurosci. Lett 2012, 513, 51–56. [Google Scholar]
- Jiménez-Aliaga, K.; Bermejo-Bescós, P.; Benedí, J.; Martín-Aragón, S. Quercetin and rutin exhibit antiamyloidogenic and fibril-disaggregating effects in vitro and potent antioxidant activity in APPswe cells. Life Sci 2011, 89, 939–945. [Google Scholar]
- Wang, S.W.; Wang, Y.J.; Su, Y.J.; Zhou, W.W.; Yang, S.G.; Zhang, R.; Zhao, M.; Li, Y.N.; Zhang, Z.P.; Zhan, D.W.; et al. Rutin inhibits β-amyloid aggregation and cytotoxicity, attenuates oxidative stress, and decreases the production of nitric oxide and proinflammatory cytokines. Neurotoxicology 2012, 33, 482–490. [Google Scholar]
- Liu, R.; Meng, F.; Zhang, L.; Liu, A.; Qin, H.; Lan, X.; Li, L.; Du, G. Luteolin isolated from the medicinal plant Elsholtzia rugulosa (Labiatae) prevents copper-mediated toxicity in β-amyloid precursor protein Swedish mutation overexpressing SH-SY5Y cells. Molecules 2011, 16, 2084–2096. [Google Scholar]
- Sarkar, N.; Kumar, M.; Dubey, V.K. Rottlerin dissolves pre-formed protein amyloid: A study on hen egg white lysozyme. Biochim. Biophys. Acta 2011, 1810, 809–814. [Google Scholar]
- Maioli, E.; Torricelli, C.; Valacchi, G. Rottlerin and curcumin: A comparative analysis. Ann. N. Y. Acad. Sci 2012, 1259, 65–76. [Google Scholar]
- Rivière, C.; Richard, T.; Vitrac, X.; Mérillon, J.M.; Valls, J.; Monti, J.P. New polyphenols active on beta-amyloid aggregation. Bioorg. Med. Chem. Lett 2008, 18, 828–831. [Google Scholar]
- Bastianetto, S.; Dumont, Y.; Han, Y.; Quirion, R. Comparative neuroprotective properties of stilbene and catechin analogs: Action via a plasma membrane receptor site? CNS Neurosci. Ther 2009, 15, 76–83. [Google Scholar]
- Richard, T.; Poupard, P.; Nassra, M.; Papastamoulis, Y.; Iglésias, M.L.; Krisa, S.; Waffo-Teguo, P.; Mérillon, J.M.; Monti, J.P. Protective effect of ɛ-viniferin on β-amyloid peptide aggregation investigated by electrospray ionization mass spectrometry. Bioorg. Med. Chem 2011, 19, 3152–3155. [Google Scholar]
- Ono, K.; Hirohata, M.; Yamada, M. Ferulic acid destabilizes preformed beta-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun 2005, 336, 444–449. [Google Scholar]
- Sgarbossa, A.; Monti, S.; Lenci, F.; Bramanti, E.; Bizzarri, R.; Barone, V. The effects of ferulic acid on β-amyloid fibrillar structures investigated through experimental and computational techniques. Biochim. Biophys. Acta 2013, 1830, 2924–2937. [Google Scholar]
- Yan, J.J.; Jung, J.S.; Kim, T.K.; Hasan, A.; Hong, C.W.; Nam, J.S.; Song, D.K. Protective effects of ferulic acid in amyloid precursor protein plus presenilin-1 transgenic mouse model of Alzheimer disease. Biol. Pharm. Bull 2013, 36, 140–143. [Google Scholar]





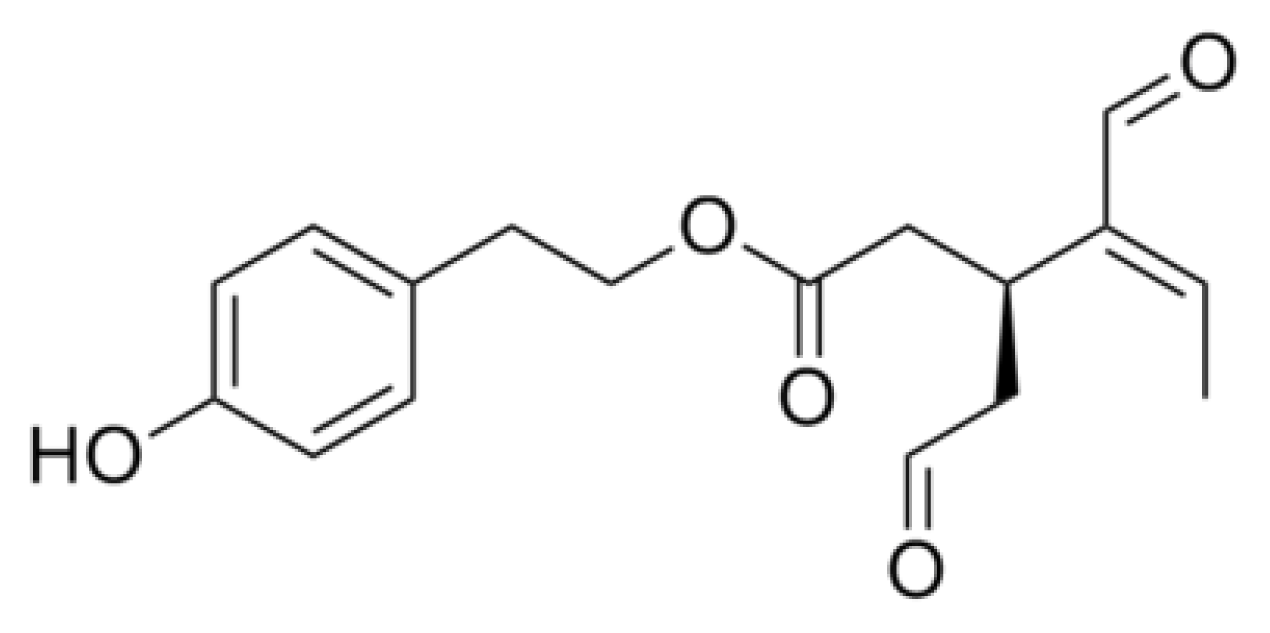

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stefani, M.; Rigacci, S. Protein Folding and Aggregation into Amyloid: The Interference by Natural Phenolic Compounds. Int. J. Mol. Sci. 2013, 14, 12411-12457. https://doi.org/10.3390/ijms140612411
Stefani M, Rigacci S. Protein Folding and Aggregation into Amyloid: The Interference by Natural Phenolic Compounds. International Journal of Molecular Sciences. 2013; 14(6):12411-12457. https://doi.org/10.3390/ijms140612411
Chicago/Turabian StyleStefani, Massimo, and Stefania Rigacci. 2013. "Protein Folding and Aggregation into Amyloid: The Interference by Natural Phenolic Compounds" International Journal of Molecular Sciences 14, no. 6: 12411-12457. https://doi.org/10.3390/ijms140612411