MAEWEST Expression in Flower Development of Two Petunia Species


 and
and
Abstract
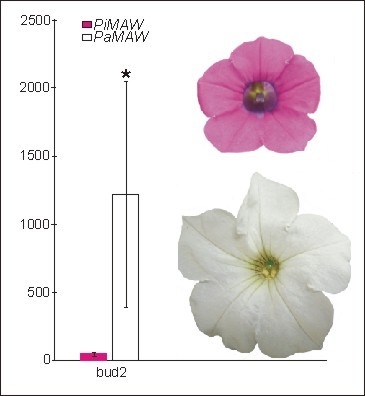
:
1. Introduction
2. Results
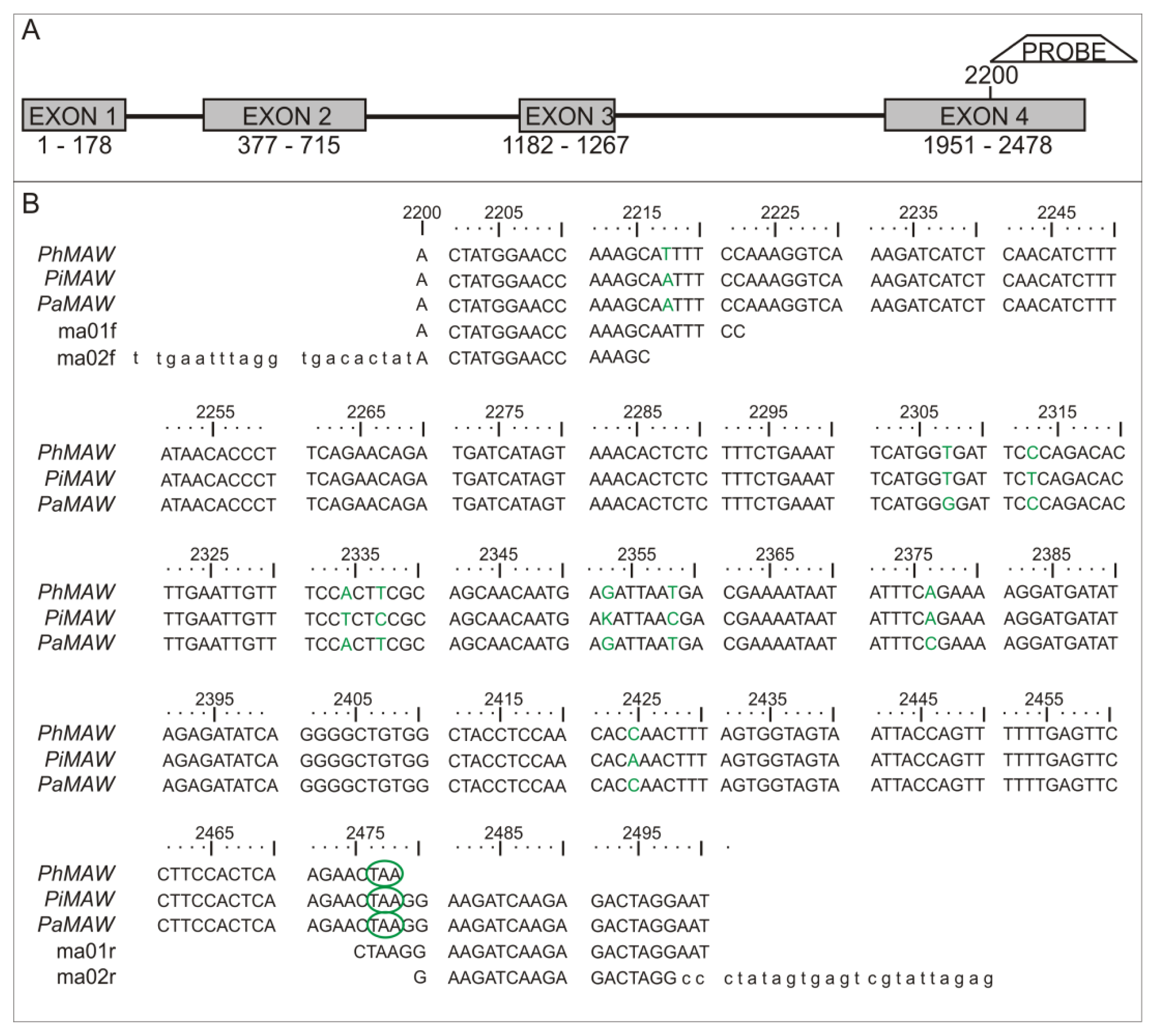
2.1. Sequence Analysis
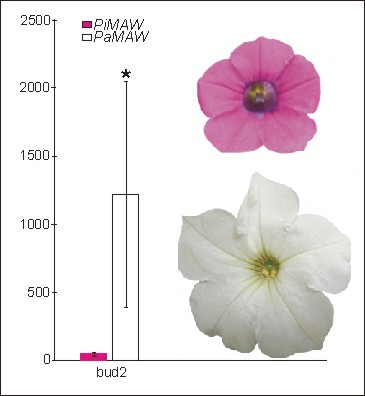
2.2. RT-PCR and qRT-PCR
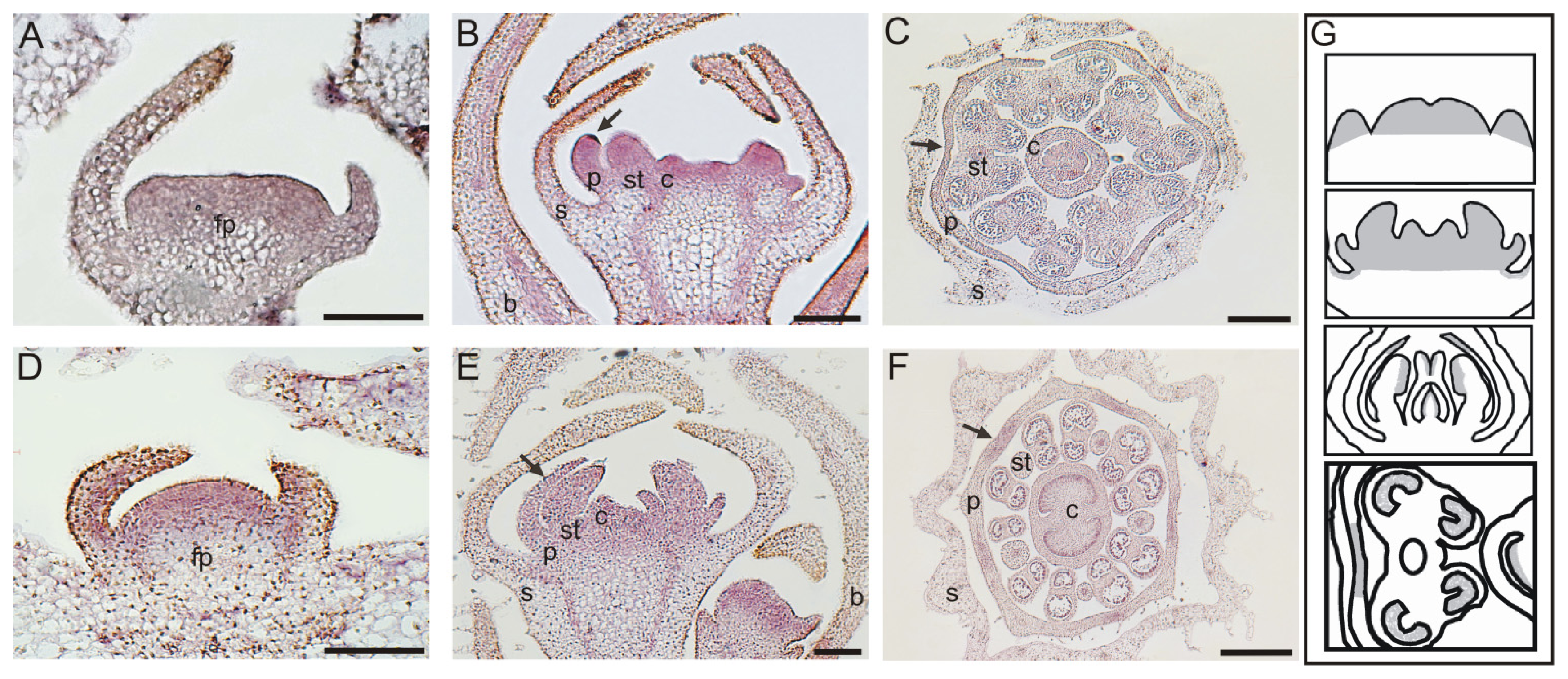
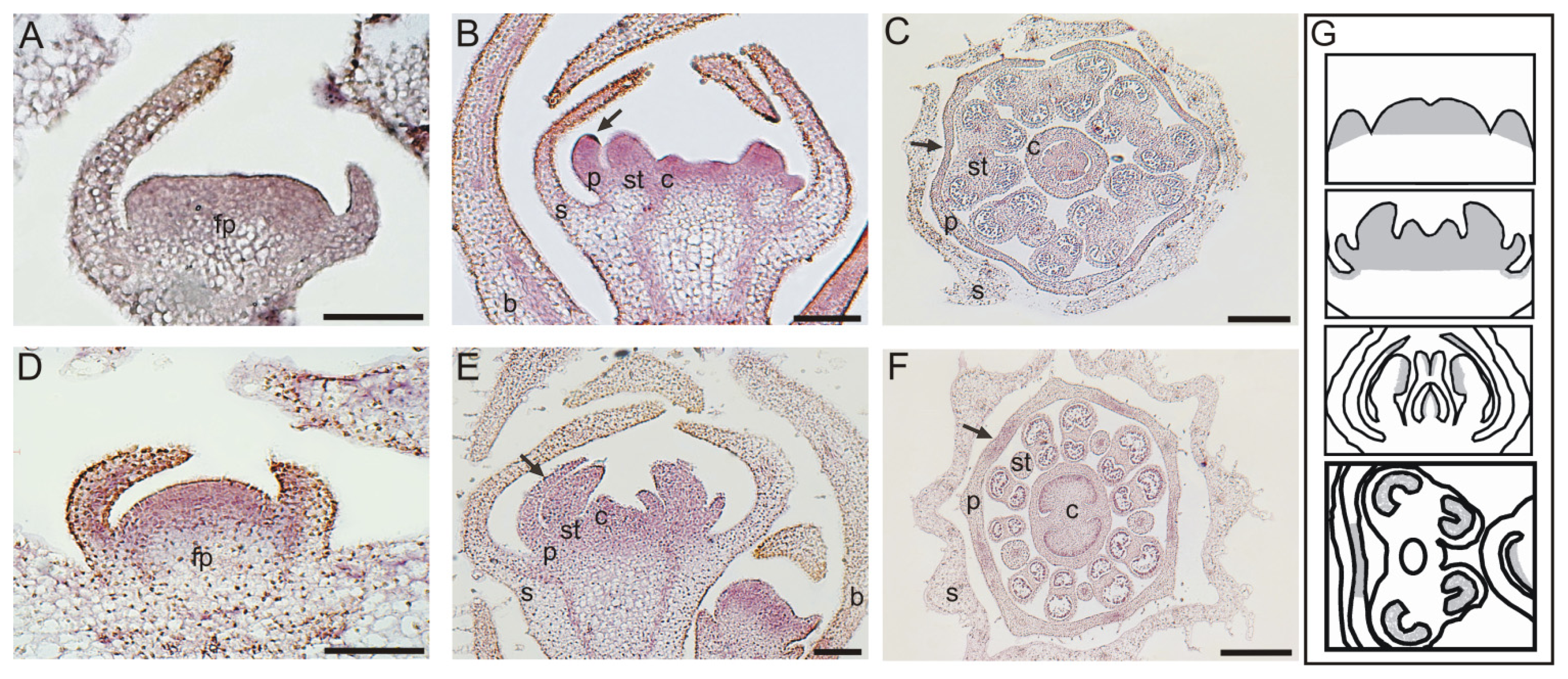
2.3. In Situ Hybridization
3. Discussion
4. Experimental Section
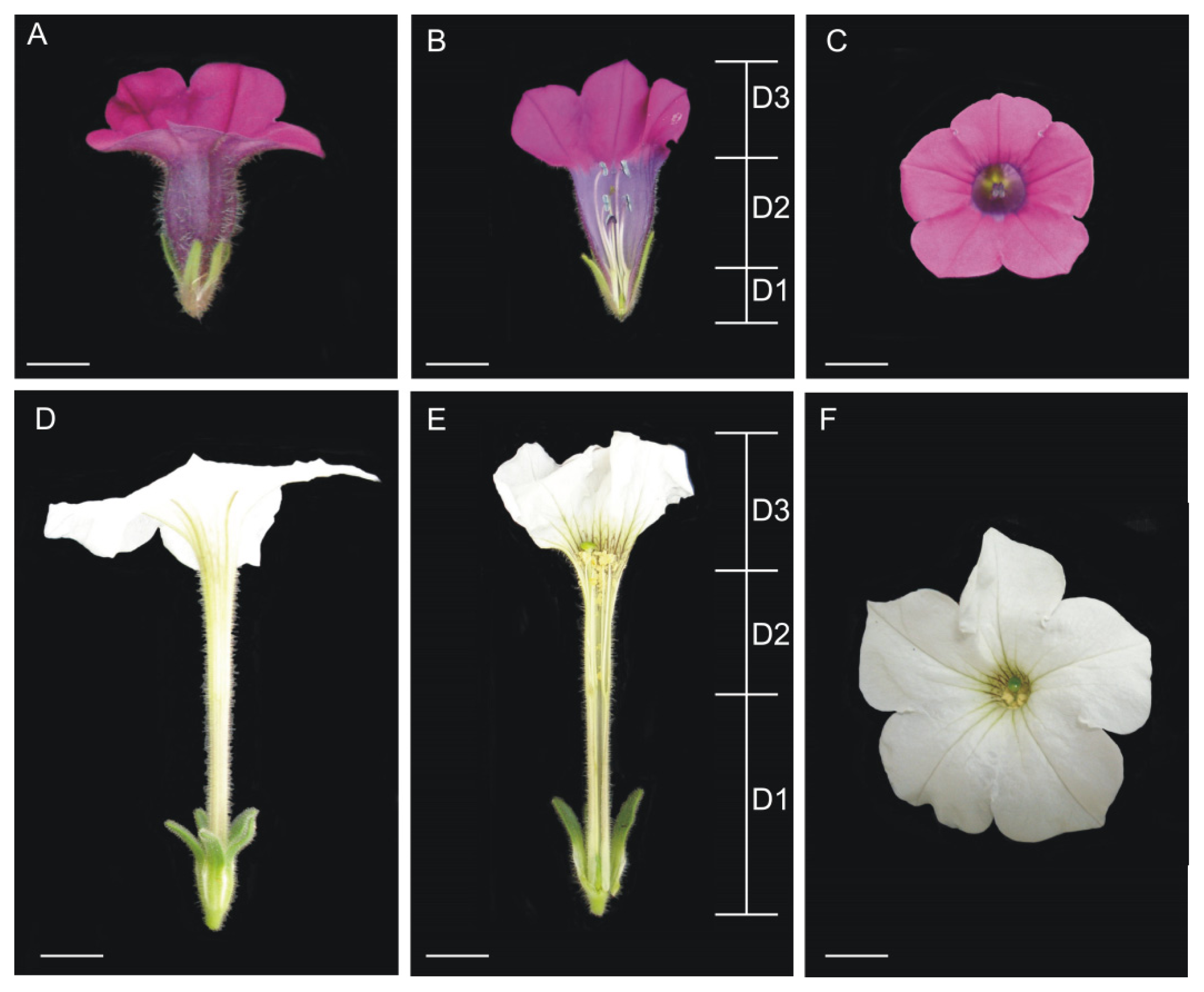
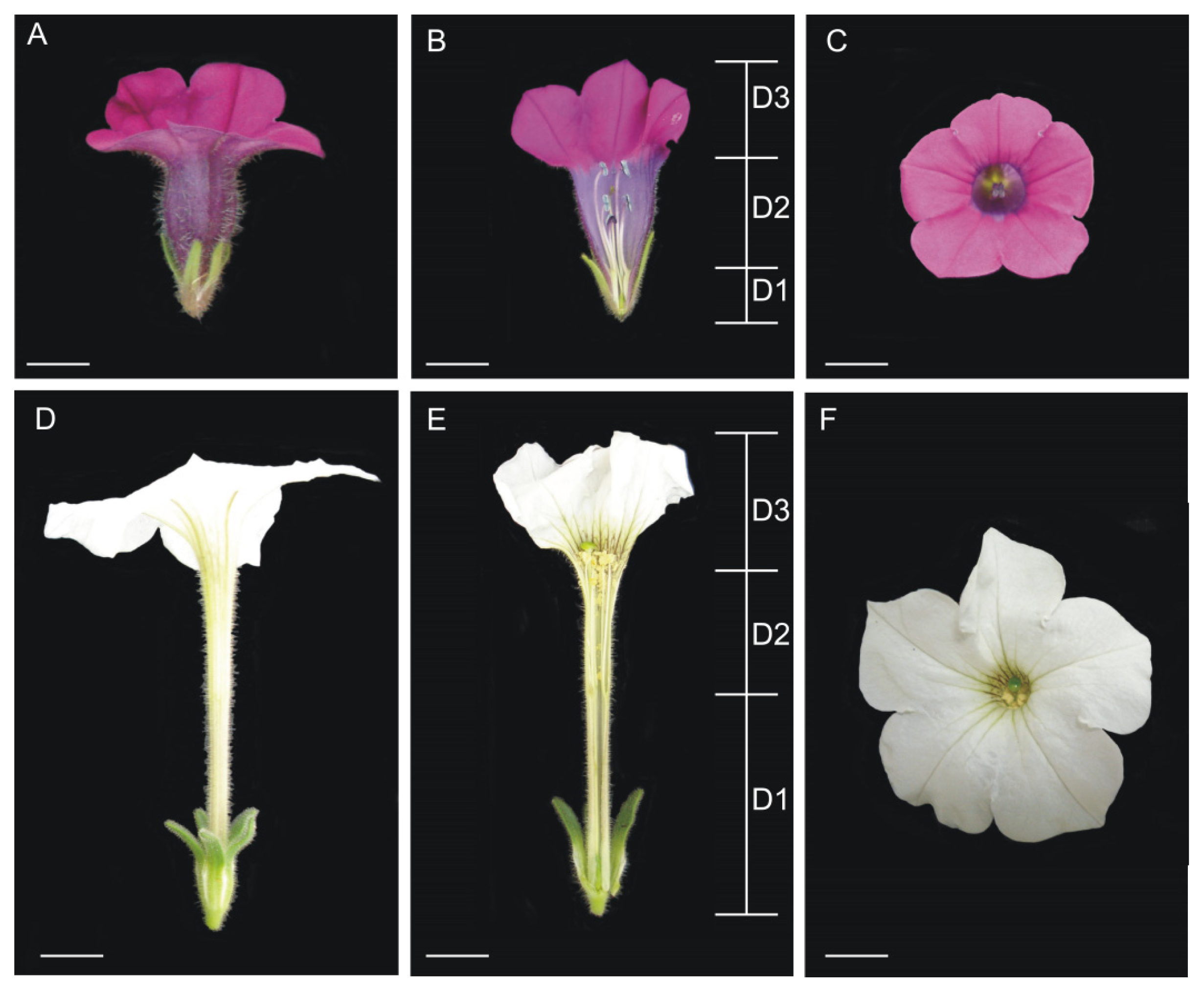
4.1. Plant Material
4.2. Sequencing and Riboprobe Labeling
4.3. RNA Extraction, RT-PCR and qRT-PCR
4.4. In Situ Hybridization
5. Conclusions
Supplementary Information
ijms-14-13796-s001.pdfAcknowledgments
Conflict of Interest
References
- Schiestl, F.P.; Schlüter, P.M. Floral isolation, specialized pollination, and pollinator behavior in orchids. Annu. Rev. Entomol 2009, 54, 425–446. [Google Scholar]
- Schemske, D.W.; Bradshaw, H.D. Pollinator preference and the evolution of floral traits in monkeyflowers (Mimulus). Proc. Natl. Acad. Sci. USA 1999, 96, 11910–11915. [Google Scholar]
- Klahre, U.; Gurba, A.; Hermann, K.; Saxenhofer, M.; Bossolini, E.; Guerin, P.M.; Kuhlemeier, C. Pollinator choice in Petunia depends on two major genetic loci for floral scent production. Curr. Biol 2011, 21, 1–10. [Google Scholar]
- Angenent, G.C.; Franken, J.; Busscher, M.; van Dijken, A.; van Went, J.L.; Dons, H.J.M.; van Tunen, A.J. A novel class of MADS box genes is lnvolved in ovule development in Petunia. Plant Cell 1995, 7, 1569–1582. [Google Scholar]
- Stuurman, J.; Hoballah, M.E.; Broger, L.; Moore, J.; Basten, C.; Kuhlemeier, C. Dissection of floral pollination syndromes in petunia. Genetics 2004, 168, 1585–1599. [Google Scholar]
- Stehmann, J.R.; Lorenz-Lemke, A.P.; Freitas, L.B.; Semir, J. The Genus. In Petunia Petunia: Evolutionary, Developmental and Physiological Genetics, 2nd ed; Gerats, T., Strommer, J., Eds.; Springer: New York, NY, USA, 2009; pp. 1–28. [Google Scholar]
- Vandenbussche, M.; Horstman, A.; Zethof, J.; Koes, R.; Rijpkema, A.S.; Gerats, T. Differential recruitment of WOX transcription factors for lateral development and organ fusion in Petunia and Arabidopsis. Plant Cell 2009, 21, 2269–2283. [Google Scholar]
- Shimizu, R.; Ji, J.; Kelsey, E.; Ohtsu, K.; Schnable, P.S.; Scanlon, M.J. Tissue specificity and evolution of meristematic WOX3 function. Plant Physiol 2009, 149, 841–850. [Google Scholar]
- Matsumoto, N.; Okada, K. A homeobox gene, PRESSED FLOWER, regulates lateral axis-dependent development of Arabidopsis flowers. Genes Dev 2001, 15, 3355–3364. [Google Scholar]
- Haecker, A.; Gross-Hardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657–668. [Google Scholar]
- Nardmann, J.; Ji, J.; Werr, W.; Scanlon, M.J. The maize duplicate genes narrow sheath1 and narrow sheath2 encode a conserved homeobox gene function in a lateral domain of shoot apical meristems. Development 2004, 131, 2827–2839. [Google Scholar]
- Wijsman, H.J.W. On the interrelationships of certain species of Petunia. II. Experimental data: Crosses between different taxa. Acta Bot. Neerl 1983, 32, 97–107. [Google Scholar]
- Ando, T.; Nomura, M.; Tsukahara, J.; Watanabe, H.; Kokubun, H.; Tsukamoto, T.; Hashimoto, G.; Marchesi, E.; Kitching, I.J. Reproductive isolation in a native population of Petunia sensu Jussieu (Solanaceae). Ann. Bot 2001, 88, 403–413. [Google Scholar]
- Gübitz, T.; Hoballah, M.E.; Dell’Olivo, A.; Kuhlemeier, C. Petunia as a Model System for the Genetics and Evolution of Pollination Syndromes. In Petunia: Evolutionary, Developmental and Physiological Genetics, 2nd ed; Gerats, T., Strommer, J., Eds.; Springer: New York, NY, USA, 2009; pp. 29–49. [Google Scholar]
- BLASTX. Available online: http://blast.ncbi.nlm.nih.gov (on accessed 26 January 2013).
- Rieu, I.; Powers, S.J. Real-time quantitative RT-PCR: Design, calculations, and statistics. Plant Cell 2009, 21, 1031–1033. [Google Scholar]
- Hoballah, M.H.; Gübitz, T.; Stuurman, J.; Broger, L.; Barone, M.; Mandel, T.; Dell’Olivo, A.; Arnold, M.; Kuhlemeier, C. Single gene-mediated shift in pollinator attraction in Petunia. Plant Cell 2007, 19, 779–790. [Google Scholar]
- Venail, J.; Dell’Olivo, A.; Kuhlemeier, C. Speciation genes in the genus Petunia. Phil. Trans. R Soc. B 2010, 365, 461–468. [Google Scholar]
- Reale, L.; Porceddu, A.; Moretti, L.L.C.; Zenoni, S.; Pezzotti, M.; Romano, B.; Ferranti, F. Patterns of cell division and expansion in developing petals of Petunia hybrida. Sex Plant Reprod 2002, 15, 123–132. [Google Scholar]
- Tadege, M.; Lin, H.; Bedair, M.; Berbel, A.; Wen, J.; Rojas, C.M.; Niu, L.; Tang, Y.; Sumner, L.; Ratet, P.; et al. STENOFOLIA regulates blade outgrowth and leaf vascular patterning in Medicago truncatula and Nicotiana sylvestris. Plant Cell 2011, 23, 2125–2142. [Google Scholar]
- Zhuang, L.; Ambrose, M.; Rameau, C.; Weng, L.; Yang, J.; Hu, X.; Luo, D.; Li, X. LATHYROIDES, encoding a WUSCHEL-related homeobox1 transcription factor, controls organ lateral growth, and regulates tendril and dorsal petal identities in garden pea (Pisum sativum L.). Mol. Plant 2012, 6, 1333–1345. [Google Scholar]
- Lin, H.; Niu, L.; McHale, N.A.; Ohme-Takagi, M.; Mysore, K.S.; Tadege, M. Evolutionarily conserved repressive activity of WOX proteins mediates leaf blade outgrowth and floral organ development in plants. Proc. Natl. Acad. Sci. USA 2013, 110, 366–371. [Google Scholar]
- Roy, A.; Frascaria, N.; MacKay, J.; Bousquet, J. Segregating random amplified polymorphic DNAs (RAPDs) in Betula alleghaniensis. Theor. Appl. Genet 1992, 85, 173–180. [Google Scholar]
- Petunia 454 database. Available online: http://biosrv.cab.unina.it/454petuniadb/ (on accessed 5 May 2010).
- Rozen, S.; Skaletsky, H.J. Primer3 on the WWW for General Users and for Biologist Programmers. In Bioinformatics Methods and Protocols: Methods in Molecular Biology; Krawetz, S., Misener, S., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar]
- Dunn, I.S.; Blattner, F.R. Charons 36 to 40: Multi-enzyme, high capacity, recombination deficient replacement vectors with polylinkers and polystuffers. Nucleic Acids Res 1987, 15, 2677–2698. [Google Scholar]
- GenBank. Available online: http://www.ncbi.nlm.nih.gov (on accessed 6 June 2013).
- Phytozome. Available online: http://www.phytozome.net (on accessed 6 June 2013).
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol 2011, 28, 2731–2739. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar]
- Dornelas, M.C.; van Lammeren, A.A.M.; Kreis, M. Arabidopsis thaliana SHAGGY-related protein kinases (AtSK11 and 12) function in perianth and gynoecium development. Plant J 2000, 21, 419–429. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Petunia axillaris | Petunia inflata |
|---|---|---|
| Pollen | yellow | bluish |
| Petal color | white | purple |
| Filaments | adnated to the middle of the tube | adnated to the base of the tube |
| Corolla shape | hypocrateriform | funnelform |
| Self-compatibility | self-compatible/self-incompatible | self-incompatible |
| Nectar | large amounts | low amounts |
| Volatiles | large amounts | low amounts |
| Habit | erect | ascendant |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Segatto, A.L.A.; Turchetto-Zolet, A.C.; Aizza, L.C.B.; Monte-Bello, C.C.; Dornelas, M.C.; Margis, R.; Freitas, L.B. MAEWEST Expression in Flower Development of Two Petunia Species. Int. J. Mol. Sci. 2013, 14, 13796-13807. https://doi.org/10.3390/ijms140713796
Segatto ALA, Turchetto-Zolet AC, Aizza LCB, Monte-Bello CC, Dornelas MC, Margis R, Freitas LB. MAEWEST Expression in Flower Development of Two Petunia Species. International Journal of Molecular Sciences. 2013; 14(7):13796-13807. https://doi.org/10.3390/ijms140713796
Chicago/Turabian StyleSegatto, Ana Lúcia A., Andreia Carina Turchetto-Zolet, Lilian Cristina B. Aizza, Carolina C. Monte-Bello, Marcelo C. Dornelas, Rogerio Margis, and Loreta B. Freitas. 2013. "MAEWEST Expression in Flower Development of Two Petunia Species" International Journal of Molecular Sciences 14, no. 7: 13796-13807. https://doi.org/10.3390/ijms140713796