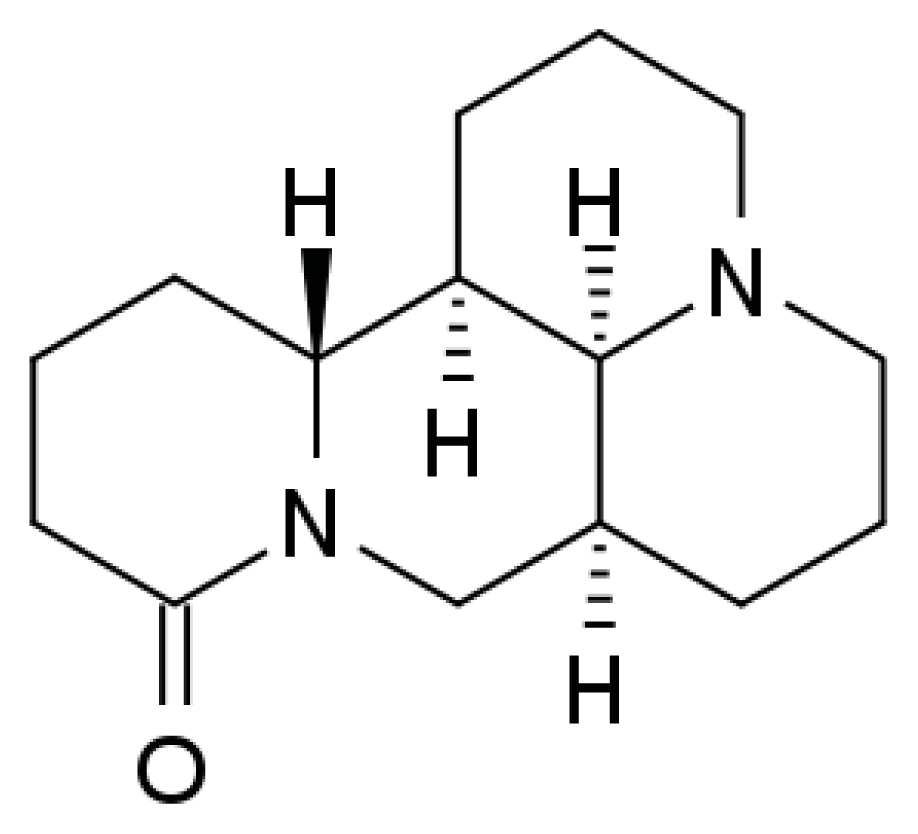
Matrine Activates PTEN to Induce Growth Inhibition and Apoptosis in V600EBRAF Harboring Melanoma Cells

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
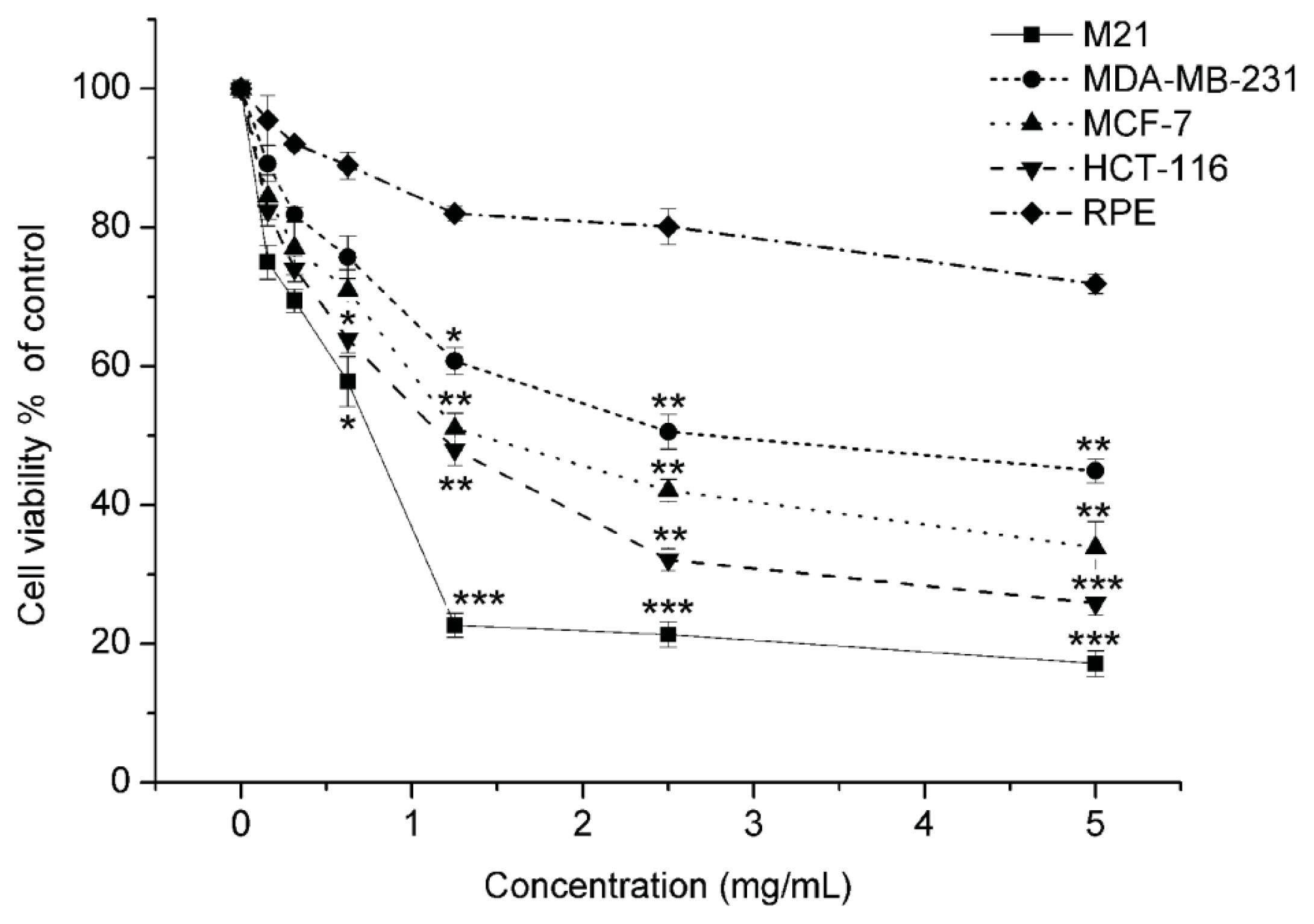
2.1.1. Matrine Exhibited Effective Proliferation Inhibition in M21 Melanoma Cells, but Did Not Affect the Normal Cells
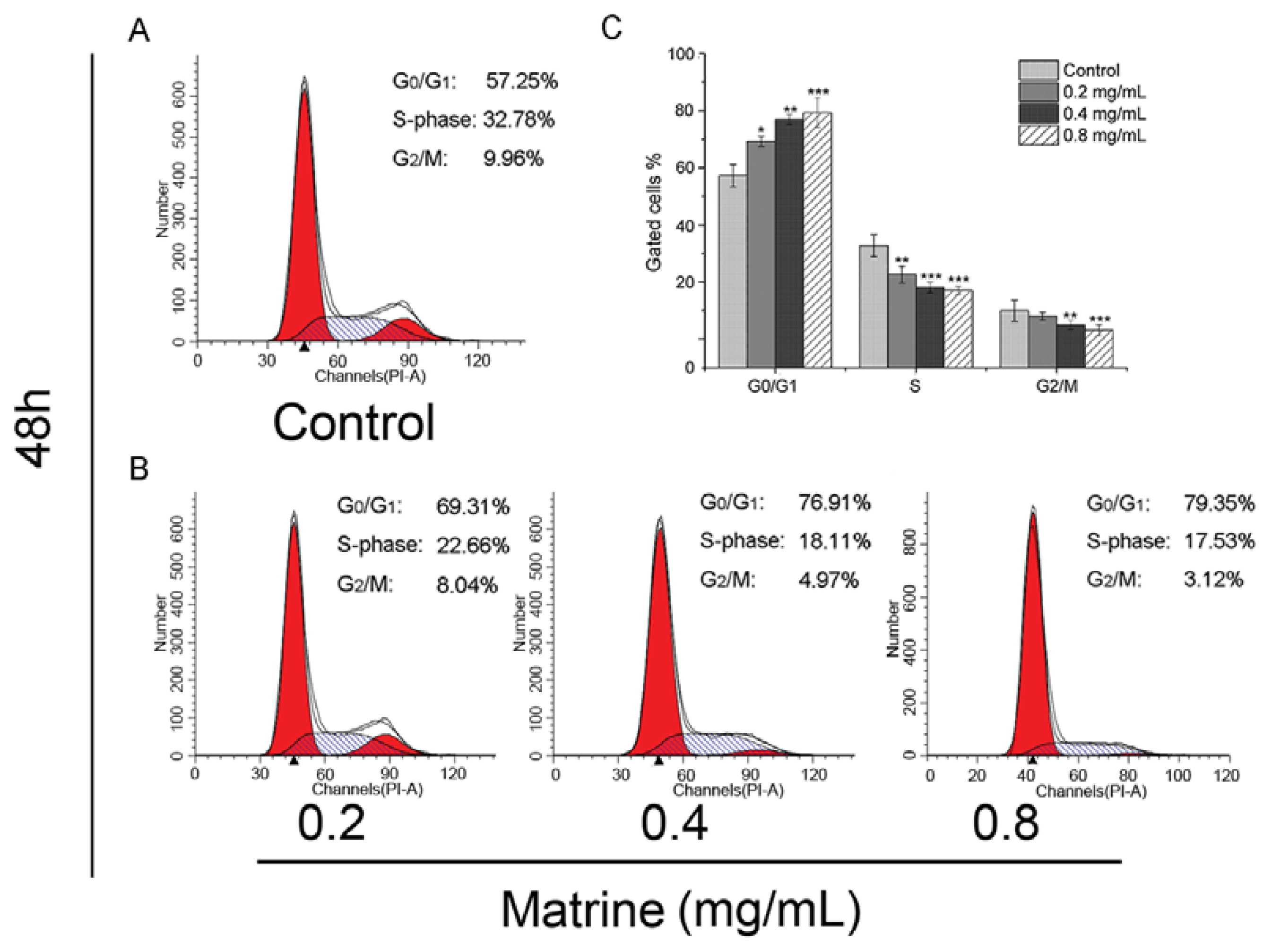
2.1.2. Matrine Induced G0/G1 Phase Cell Cycle Arrest Dose-Dependently
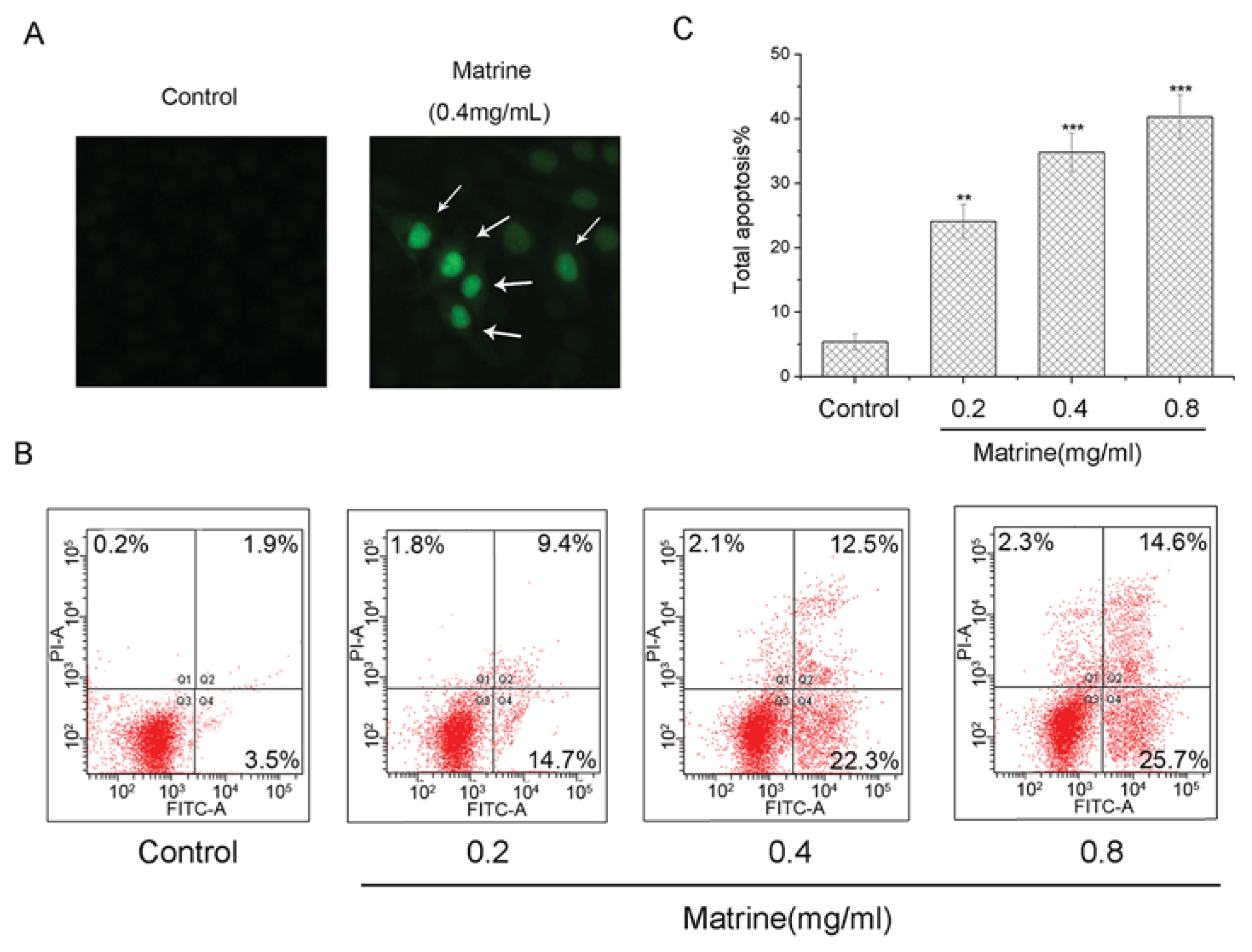
2.1.3. Matrine Induced Apoptosis in M21 Cells Dose-Dependently
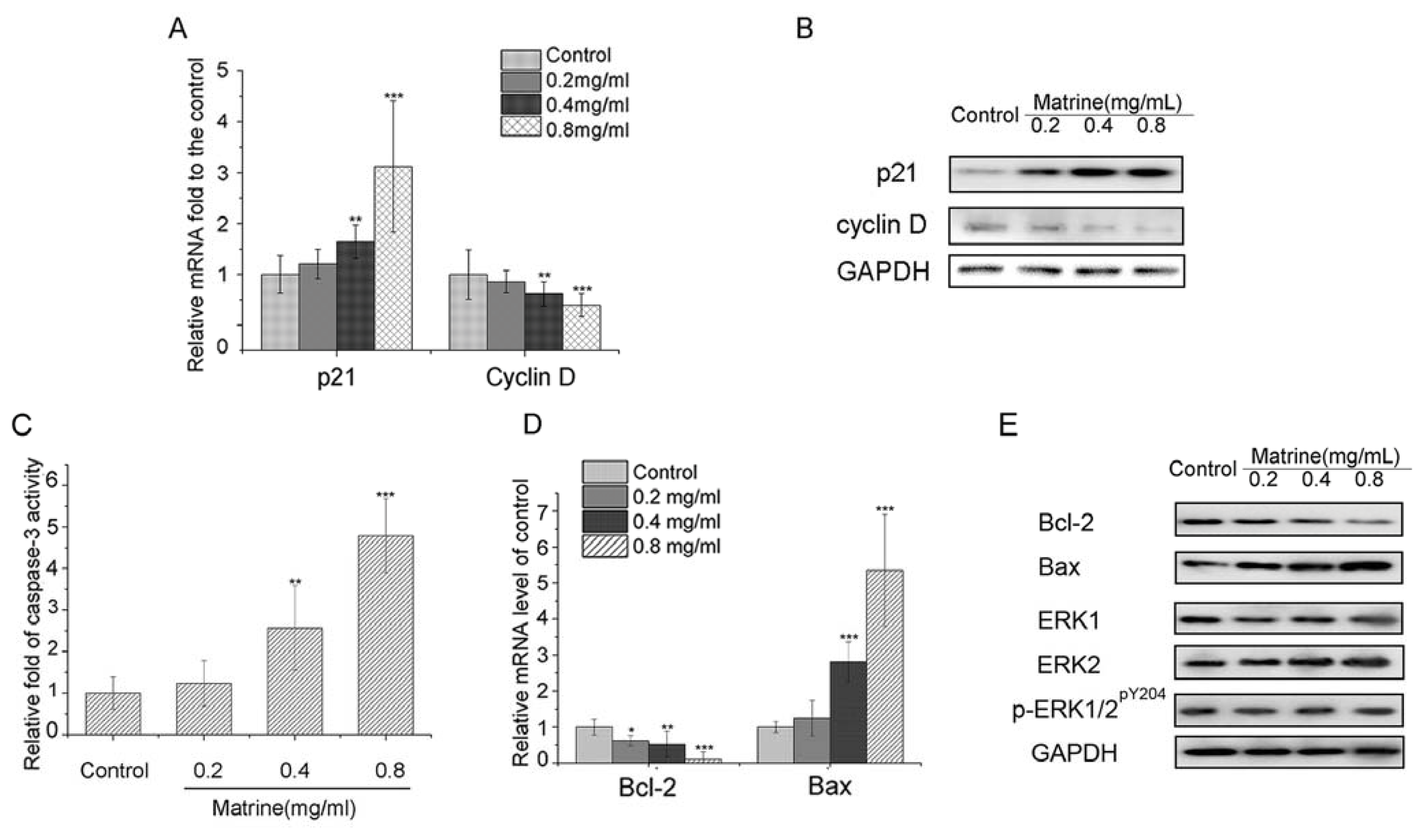
2.1.4. Matrine Upregulated p21 and Downregulated Cyclin D in M21 Cells
2.1.5. Both Caspase-3 Activation and Bcl-2/Bax Interference Involved in the Apoptosis Induced by Matrine
2.1.6. Matrine Did Not Affect the Protein Expression of ERK1/2 or Phosphorylation of ERK1/2 in M21 Cells
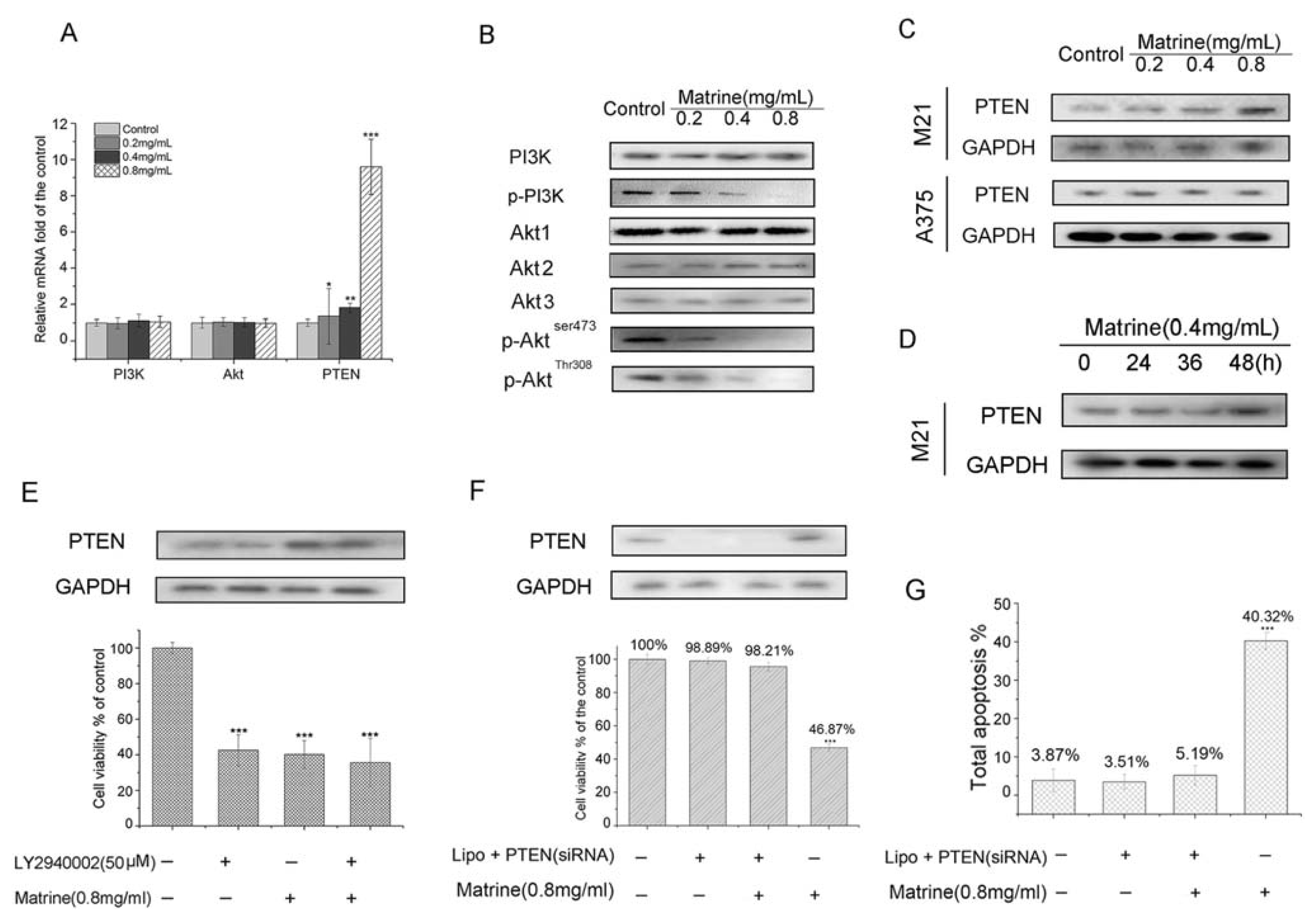
2.1.7. Matrine Activated PTEN to the Inhibit PI3K/Akt Pathway in M21 Cells, but Not in A375 Cells
2.1.8. Matrine Enhanced the Inhibition of PI3K/Akt Pathway to Inhibit Cell Proliferation in M21 Cells
2.1.9. PTEN Silencing Blocked the Cell Growth Inhibition and Apoptosis Induced by Matrine in M21 Cells
2.2. Discussion
3. Experimental Section
3.1. Chemicals
3.2. Cell Lines and Cell Culture
3.3. Cell Viability Assay
3.4. Flow Cytometric Analysis of Cellular DNA Content
3.5. Flow Cytometric Analysis of Apoptosis and Necrosis
3.6. TUNEL Apoptosis Detection Assay
3.7. Determination of Caspase Activity
3.8. Real-Time Quantitative Reverse Transcription-PCR Analysis
3.9. Western Blot Analysis
3.10. Cell Transfection
3.11. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet 1997, 15, 356–362. [Google Scholar]
- Furnari, F.B.; Huang, H.J.; Cavenee, W.K. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res 1998, 58, 5002–5008. [Google Scholar]
- Georgescu, M.M.; Kirsch, K.H.; Kaloudis, P.; Yang, H.; Pavletich, N.P.; Hanafusa, H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res 2000, 60, 7033–7038. [Google Scholar]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol 2009, 4, 127–150. [Google Scholar]
- Garcia-Echeverria, C.; Sellers, W. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 2008, 27, 5511–5526. [Google Scholar]
- Lim, Y.; Han, I.; Kwon, H.; Oh, E. Trichostatin a-induced detransformation correlates with decreased focal adhesion kinase phosphorylation at tyrosine 861 inras-transformed fibroblasts. J. Biol. Chem 2002, 277, 12735–12740. [Google Scholar]
- Bali, P.; George, P.; Cohen, P.; Tao, J.; Guo, F.; Sigua, C.; Vishvanath, A.; Scuto, A.; Annavarapu, S.; Fiskus, W.; et al. Superior activity of the combination of histone deacetylase inhibitor LAQ824 and the FLT-3 kinase inhibitor PKC412 against human acute myelogenous leukemia cells with mutant FLT-3. Clin. Cancer Res 2004, 10, 4991–4997. [Google Scholar]
- Gan, Y.H.; Zhang, S. PTEN/AKT pathway involved in histone deacetylases inhibitor induced cell growth inhibition and apoptosis of oral squamous cell carcinoma cells. Oral Oncol 2009, 45, e150–e154. [Google Scholar]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar]
- Baron, V.; Adamson, E.D.; Calogero, A.; Ragona, G.; Mercola, D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFβ1, PTEN, p53, and fibronectin. Cancer Gene Ther 2005, 13, 115–124. [Google Scholar]
- Kim, S.; Domon-Dell, C.; Kang, J.; Chung, D.H.; Freund, J.N.; Evers, B.M. Down-regulation of the tumor suppressor PTEN by the tumor necrosis factor-α/nuclear factor-κB (NF-κB)-inducing kinase/NF-κB pathway is linked to a default IκB-α autoregulatory loop. J. Biol. Chem 2004, 279, 4285–4291. [Google Scholar]
- Ishola, T.A.; Kang, J.H.; Qiao, J.; Evers, B.M.; Chung, D.H. Phosphatidylinositol 3-kinase regulation of gastrin-releasing peptide-induced cell cycle progression in neuroblastoma cells. Biochim. Biophys. Acta (BBA) 2007, 1770, 927–932. [Google Scholar]
- Vredeveld, L.C.; Possik, P.A.; Smit, M.A.; Meissl, K.; Michaloglou, C.; Horlings, H.M.; Ajouaou, A.; Kortman, P.C.; Dankort, D.; McMahon, M.; et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012, 26, 1055–1069. [Google Scholar]
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Yamanoshita, O.; Iida, M.; Ohgami, N.; Tamura, H.; Kawamoto, Y.; et al. RAS/RAF/MEK/ERK and PI3K/PTEN/AKT signaling in malignant melanoma progression and therapy. Dermatol. Res. Pract. 2012, 354191:1–354191:5. [Google Scholar]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin 2009, 59, 225–249. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Xing, F.; Persaud, Y.; Pratilas, C.A.; Taylor, B.S.; Janakiraman, M.; She, Q.B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring V600EBRAF. Oncogene 2012, 31, 446–457. [Google Scholar]
- Daud, A.; Bastian, B.C. Beyond BRAF in melanoma. Curr. Top. Microbiol. Immunol 2012, 355, 99–117. [Google Scholar]
- Mehnert, J.M.; Kluger, H.M. Driver mutations in melanoma: Lessons learned from bench-to-bedside studies. Curr. Oncol. Rep 2012, 14, 449–457. [Google Scholar]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol 2012, 13, 283–296. [Google Scholar]
- Jiang, H.; Hou, C.; Zhang, S.; Xie, H.; Zhou, W.; Jin, Q.; Cheng, X.; Qian, R.; Zhang, X. Matrine upregulates the cell cycle protein E2F-1 and triggers apoptosis via the mitochondrial pathway in K562 cells. Eur. J. Pharmacol 2007, 559, 98–108. [Google Scholar]
- Yang, Y.; Xiu, J.; Zhang, X.; Zhang, L.; Yan, K.; Qin, C.; Liu, J. Antiviral effect of matrine against human enterovirus 71. Molecules 2012, 17, 10370–10376. [Google Scholar]
- Suo, Z.; Liu, Y.; Ferreri, M.; Zhang, T.; Liu, Z.; Mu, X.; Han, B. Impact of matrine on inflammation related factors in rat intestinal microvascular endothelial cells. J. Ethnopharmacol 2009, 125, 404–409. [Google Scholar]
- Gao, H.Y.; Li, G.Y.; Lou, M.M.; Li, X.Y.; Wei, X.Y.; Wang, J.H. Hepatoprotective effect of Matrine salvianolic acid B salt on carbon tetrachloride-induced hepatic fibrosis. J. Inflamm 2012, 9, 16, :1–16:9.. [Google Scholar]
- Li, X.; Chu, W.; Liu, J.; Xue, X.; Lu, Y.; Shan, H.; Yang, B. Antiarrhythmic properties of long-term treatment with matrine in arrhythmic rat induced by coronary ligation. Biol. Pharm. Bull 2009, 32, 1521–1526. [Google Scholar]
- Zheng, H.; Chen, G.; Shi, L.; Lou, Z.; Chen, F.; Hu, J. Determination of oxymatrine and its metabolite matrine in rat blood and dermal microdialysates by high throughput liquid chromatography/tandem mass spectrometry. J. Pharm. Biomed. Anal 2009, 49, 427–433. [Google Scholar]
- Dai, Z.J.; Gao, J.; Ji, Z.Z.; Wang, X.J.; Ren, H.T.; Liu, X.X.; Wu, W.Y.; Kang, H.F.; Guan, H.T. Matrine induces apoptosis in gastric carcinoma cells via alteration of Fas/FasL and activation of caspase-3. J. Ethnopharmacol 2009, 123, 91–96. [Google Scholar]
- Han, Y.; Zhang, S.; Wu, J.; Yu, K.; Zhang, Y.; Yin, L.; Bi, L. Matrine induces apoptosis of human multiple myeloma cells via activation of the mitochondrial pathway. Leuk. Lymphoma 2010, 51, 1337–1346. [Google Scholar]
- Li, H.; Tan, G.; Jiang, X.; Qiao, H.; Pan, S.; Jiang, H.; Kanwar, J.R.; Sun, X. Therapeutic effects of matrine on primary and metastatic breast cancer. Am. J. Chin. Med 2010, 38, 1115–1130. [Google Scholar]
- Zhang, J.Q.; Li, Y.M.; Liu, T.; He, W.T.; Chen, Y.T.; Chen, X.H.; Li, X.; Zhou, W.C.; Yi, J.F.; Ren, Z.J. Antitumor effect of matrine in human hepatoma G2 cells by inducing apoptosis and autophagy. World J. Gastroenterol 2010, 16, 4281–4290. [Google Scholar]
- Zhang, Z.; Wang, X.; Wu, W.; Wang, J.; Wang, Y.; Wu, X.; Fei, X.; Li, S.; Zhang, J.; Dong, P.; et al. Effects of matrine on proliferation and apoptosis in gallbladder carcinoma cells (GBC-SD). Phytother. Res 2012, 26, 932–937. [Google Scholar]
- Yang, C.L.; Liu, S.S.; Ma, Y.G.; Liu, Y.Y.; Xue, Y.X.; Huang, B. The influence of intraoperative pleural perfusion with matrine-cisplatin or cisplatin on stromal cell-derived factor-1 in non-small cell lung cancer patients with subclinical pleural metastasis. Med. Oncol 2012, 29, 574–581. [Google Scholar]
- Luo, C.; Zhong, H.J.; Zhu, L.M.; Wu, X.G.; Ying, J.E.; Wang, X.H.; Lu, W.X.; Xu, Q.; Zhu, Y.L.; Huang, J. Inhibition of matrine against gastric cancer cell line MNK45 growth and its anti-tumor mechanism. Mol. Biol. Rep 2012, 39, 5459–5464. [Google Scholar]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med 2005, 353, 2135–2147. [Google Scholar]
- Basile, K.J.; Aplin, A.E. Downregulation of Noxa by RAF/MEK inhibition counteracts cell death response in mutant B-RAF melanoma cells. Am. J. Cancer Res 2012, 2, 726–735. [Google Scholar]
- Heneberg, P. Dabrafenib: The new inhibitor of hyperactive B-RAF kinase. Klin. Onkol 2012, 25, 333–339. [Google Scholar]
- Kim, Y.K.; Ahn, S.K.; Lee, M. Differential sensitivity of melanoma cell lines with differing B-Raf mutational status to the new oncogenic B-Raf kinase inhibitor UI-152. Cancer Lett 2012, 320, 215–224. [Google Scholar]
- Konjevic, G.; Mirjacic Martinovic, K.; Vuletic, A.; Babovic, N. In-vitro IL-2 or IFN-α-induced NKG2D and CD161 NK cell receptor expression indicates novel aspects of NK cell activation in metastatic melanoma patients. Melanoma Res 2010, 20, 459–467. [Google Scholar]
- Trepiakas, R.; Berntsen, A.; Hadrup, S.R.; Bjorn, J.; Geertsen, P.F.; Straten, P.T.; Andersen, M.H.; Pedersen, A.E.; Soleimani, A.; Lorentzen, T.; et al. Vaccination with autologous dendritic cells pulsed with multiple tumor antigens for treatment of patients with malignant melanoma: Results from a phase I/II trial. Cytotherapy 2010, 12, 721–734. [Google Scholar]
- Li, L.Q.; Li, X.L.; Wang, L.; Du, W.J.; Guo, R.; Liang, H.H.; Liu, X.; Liang, D.S.; Lu, Y.J.; Shan, H.L.; et al. Matrine inhibits breast cancer growth via miR-21/PTEN/Akt pathway in MCF-7 cells. Cell. Physiol. Biochem 2012, 30, 631–641. [Google Scholar]
- Liu, X.Y.; Fang, H.; Yang, Z.G.; Wang, X.Y.; Ruan, L.M.; Fang, D.R.; Ding, Y.G.; Wang, Y.N.; Zhang, Y.; Jiang, X.L.; et al. Matrine inhibits invasiveness and metastasis of human malignant melanoma cell line A375 in vitro. Int. J. Dermatol 2008, 47, 448–456. [Google Scholar]
- Dhawan, P.; Singh, A.B.; Ellis, D.L.; Richmond, A. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-κB and tumor progression. Cancer Res 2002, 62, 7335–7342. [Google Scholar]
- Tsao, H.; Zhang, X.; Fowlkes, K.; Haluska, F.G. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res 2000, 60, 1800–1804. [Google Scholar]
- Tsao, H.; Goel, V.; Wu, H.; Yang, G.; Haluska, F.G. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J. Invest. Dermatol 2004, 122, 337–341. [Google Scholar]
- Conde-Perez, A.; Larue, L. PTEN and melanomagenesis. Future Oncol 2012, 8, 1109–1120. [Google Scholar]
- Deng, W.; Gopal, Y.N.; Scott, A.; Chen, G.; Woodman, S.E.; Davies, M.A. Role and therapeutic potential of PI3K-mTOR signaling in de novo resistance to BRAF inhibition. Pigment Cell Melanoma Res 2012, 25, 248–258. [Google Scholar]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 2011, 71, 2750–2760. [Google Scholar]
- Wang, X.; Wang, Y.; Yu, L.; Sakakura, K.; Visus, C.; Schwab, J.H.; Ferrone, C.R.; Favoino, E.; Koya, Y.; Campoli, M.R.; et al. CSPG4 in cancer: Multiple roles. Curr. Mol. Med 2010, 10, 419–429. [Google Scholar]
- Gopal, Y.N.; Deng, W.; Woodman, S.E.; Komurov, K.; Ram, P.; Smith, P.D.; Davies, M.A. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res 2010, 70, 8736–8747. [Google Scholar]
- Liu, X.-S.; Jiang, J.; Jiao, X.-Y.; Wu, Y.-E.; Lin, J.-H. Matrine-induced apoptosis in leukemia U937 cells: Involvement of caspases activation and MAPK-independent pathways. Planta Med 2006, 72, 501–506. [Google Scholar]
- Jin, H.; Liang, L.; Liu, L.; Deng, W.; Liu, J. HDAC inhibitor DWP0016 activates p53 transcription and acetylation to inhibit cell growth in U251 glioblastoma cells. J. Cell. Biochem 2013, 114, 1498–1509. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell lines | Tissue origin | Matrine |
|---|---|---|
| IC50 ± SD (mg/mL) | ||
| M21 | Melanoma | 0.769 ± 0.28 |
| MDB-MA-231 | Breast carcinoma | 2.758 ± 0.19 |
| MCF-7 | Breast carcinoma | 1.405 ± 0.35 |
| HCT116 | Colon carcinoma | 1.242 ± 0.17 |
| RPE | Human retinal pigment epithelium | >5 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jin, H.; Sun, Y.; Wang, S.; Cheng, X. Matrine Activates PTEN to Induce Growth Inhibition and Apoptosis in V600EBRAF Harboring Melanoma Cells. Int. J. Mol. Sci. 2013, 14, 16040-16057. https://doi.org/10.3390/ijms140816040
Jin H, Sun Y, Wang S, Cheng X. Matrine Activates PTEN to Induce Growth Inhibition and Apoptosis in V600EBRAF Harboring Melanoma Cells. International Journal of Molecular Sciences. 2013; 14(8):16040-16057. https://doi.org/10.3390/ijms140816040
Chicago/Turabian StyleJin, Hui, Yu Sun, Shuiying Wang, and Xiaodong Cheng. 2013. "Matrine Activates PTEN to Induce Growth Inhibition and Apoptosis in V600EBRAF Harboring Melanoma Cells" International Journal of Molecular Sciences 14, no. 8: 16040-16057. https://doi.org/10.3390/ijms140816040