A Molecular and Chemical Perspective in Defining Melatonin Receptor Subtype Selectivity

Abstract
:1. Introduction
1.1. Melatonin and Melatonin Receptors
1.2. G Protein-Coupled Melatonin Receptors
2. Melatonin Receptor Signaling
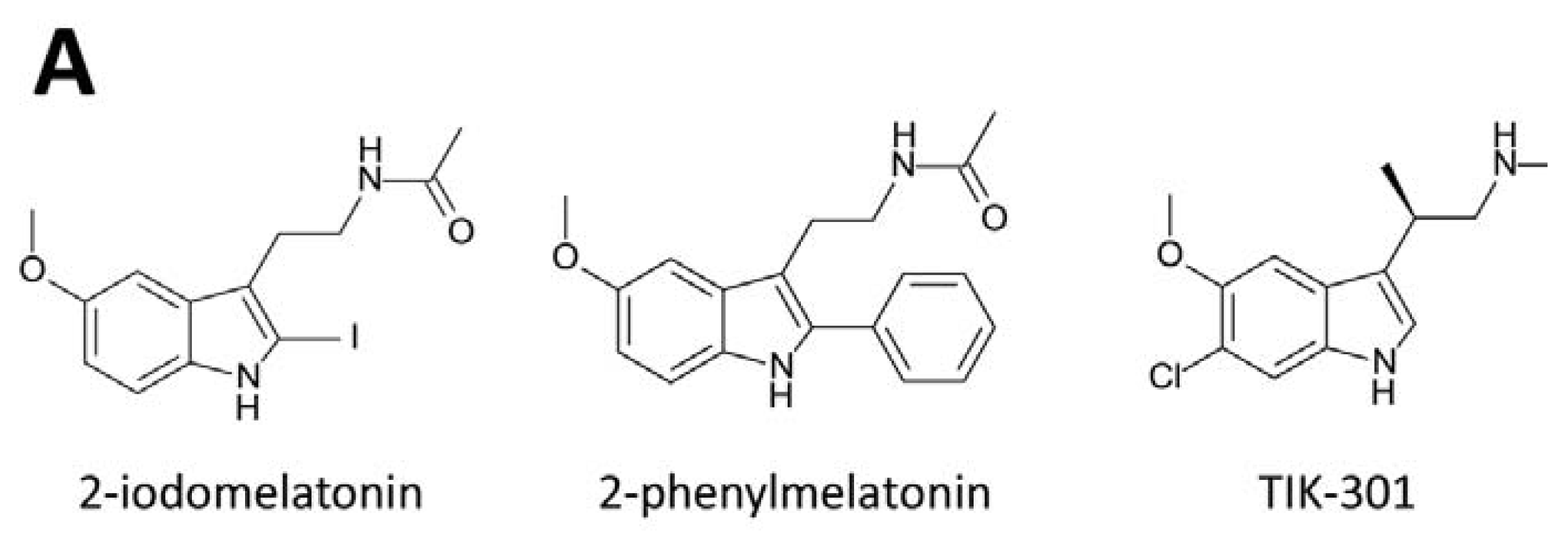
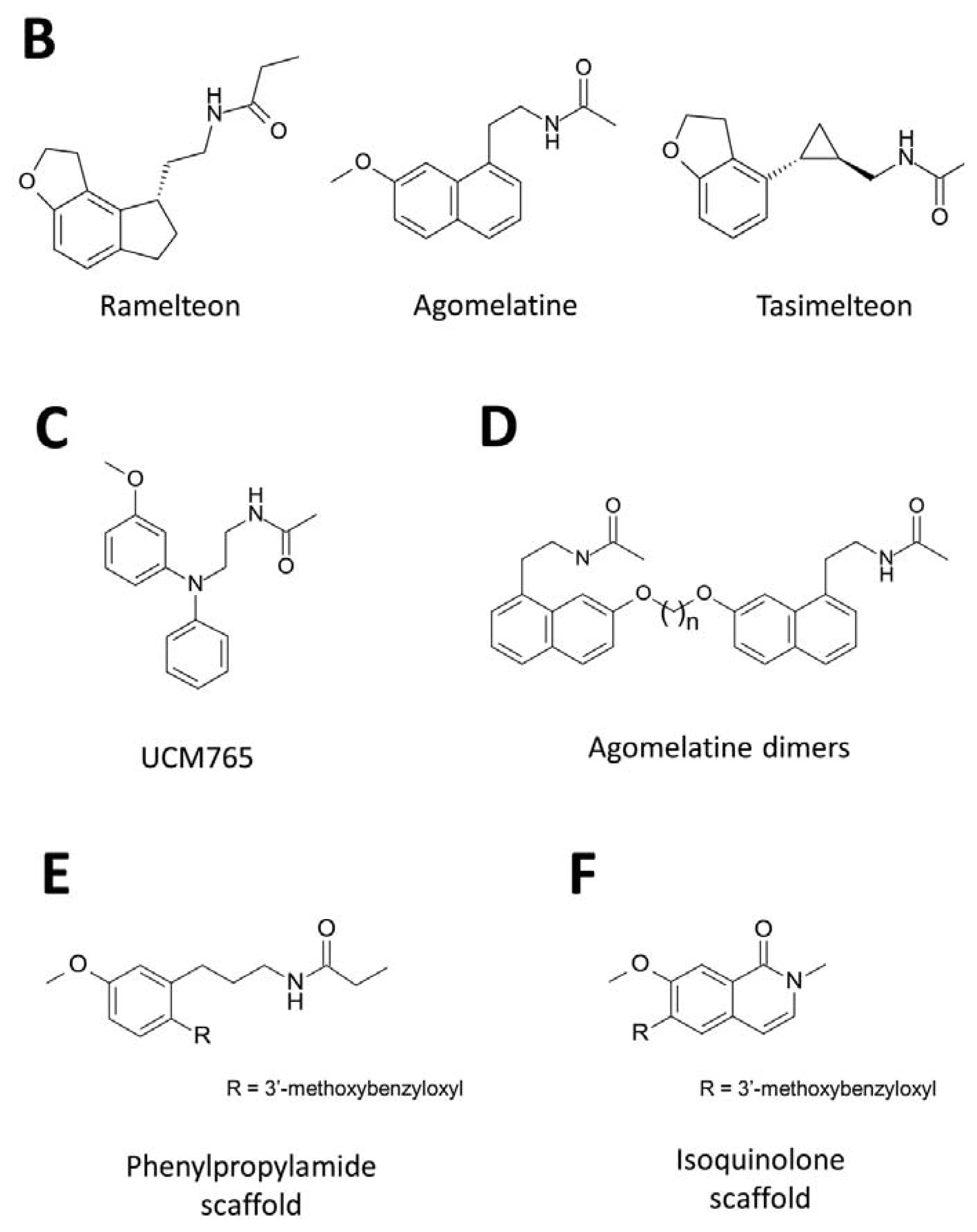
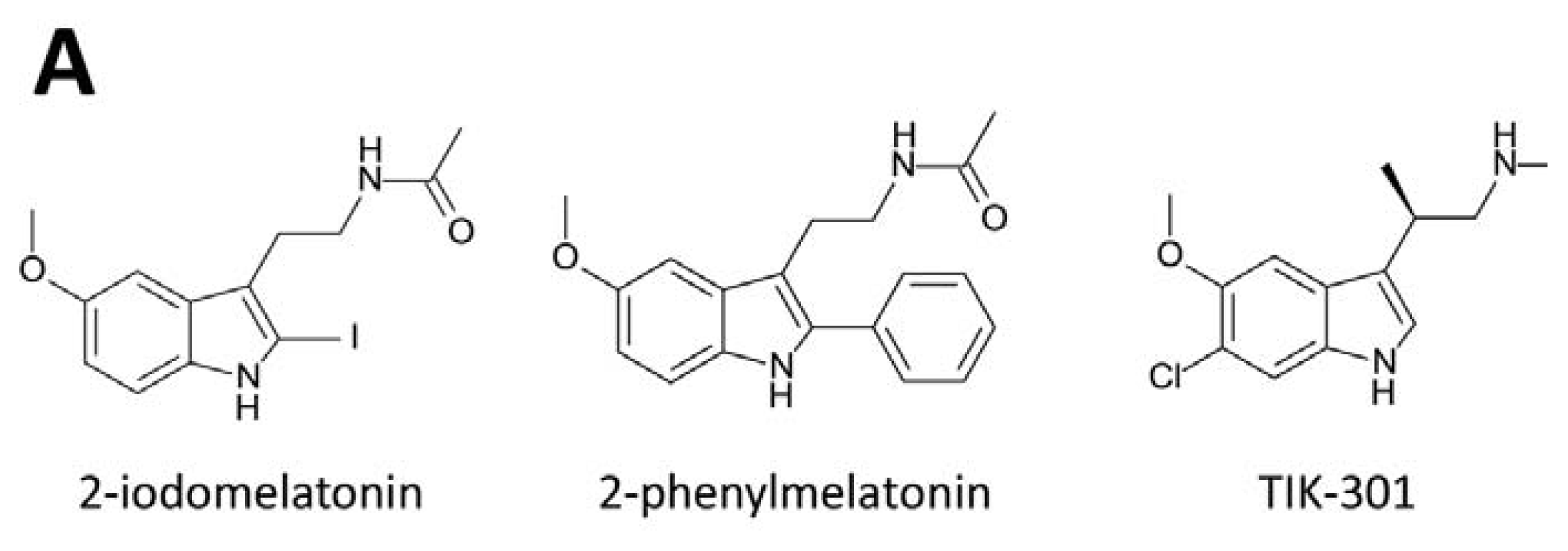
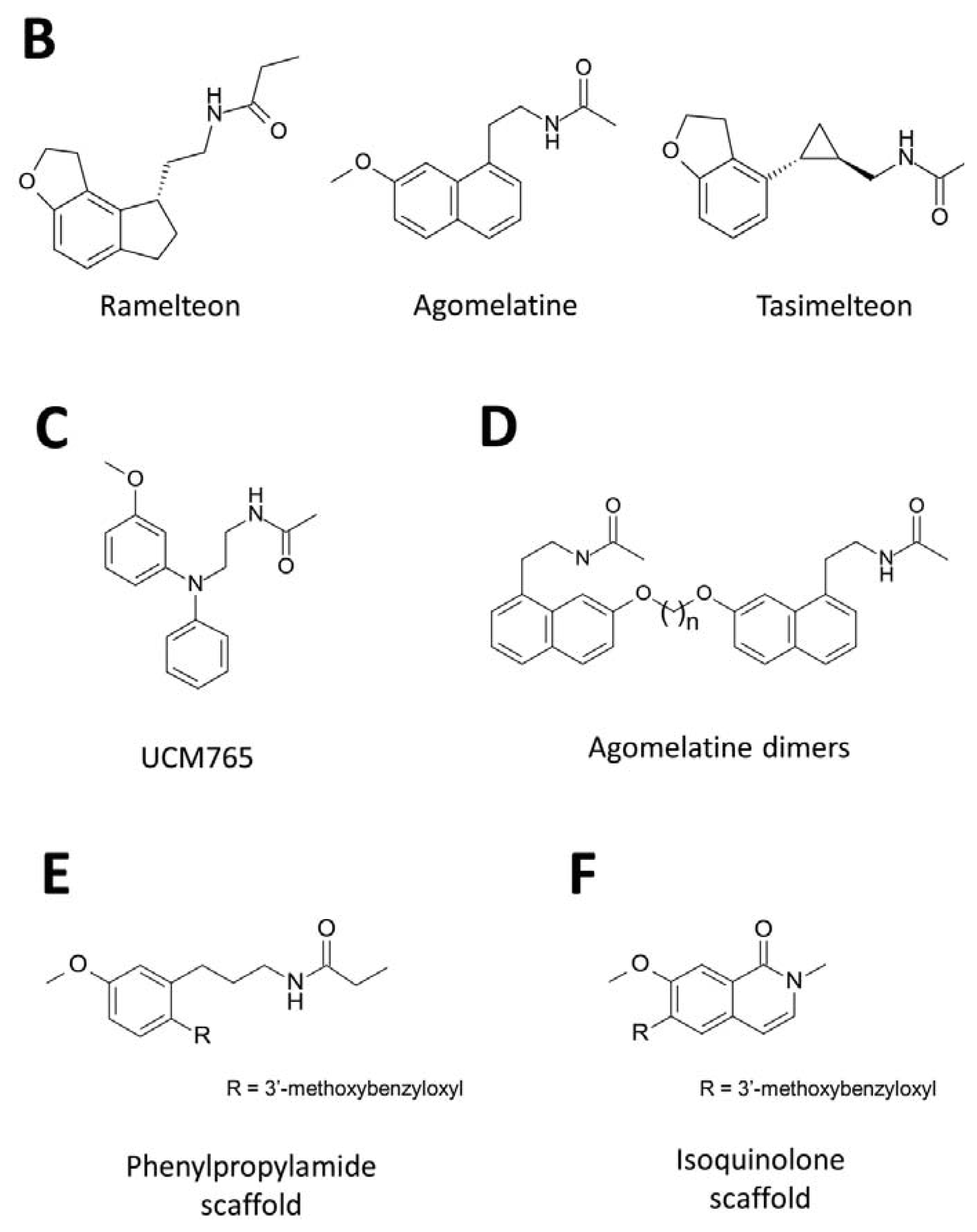
3. Development of Synthetic Melatoninergics
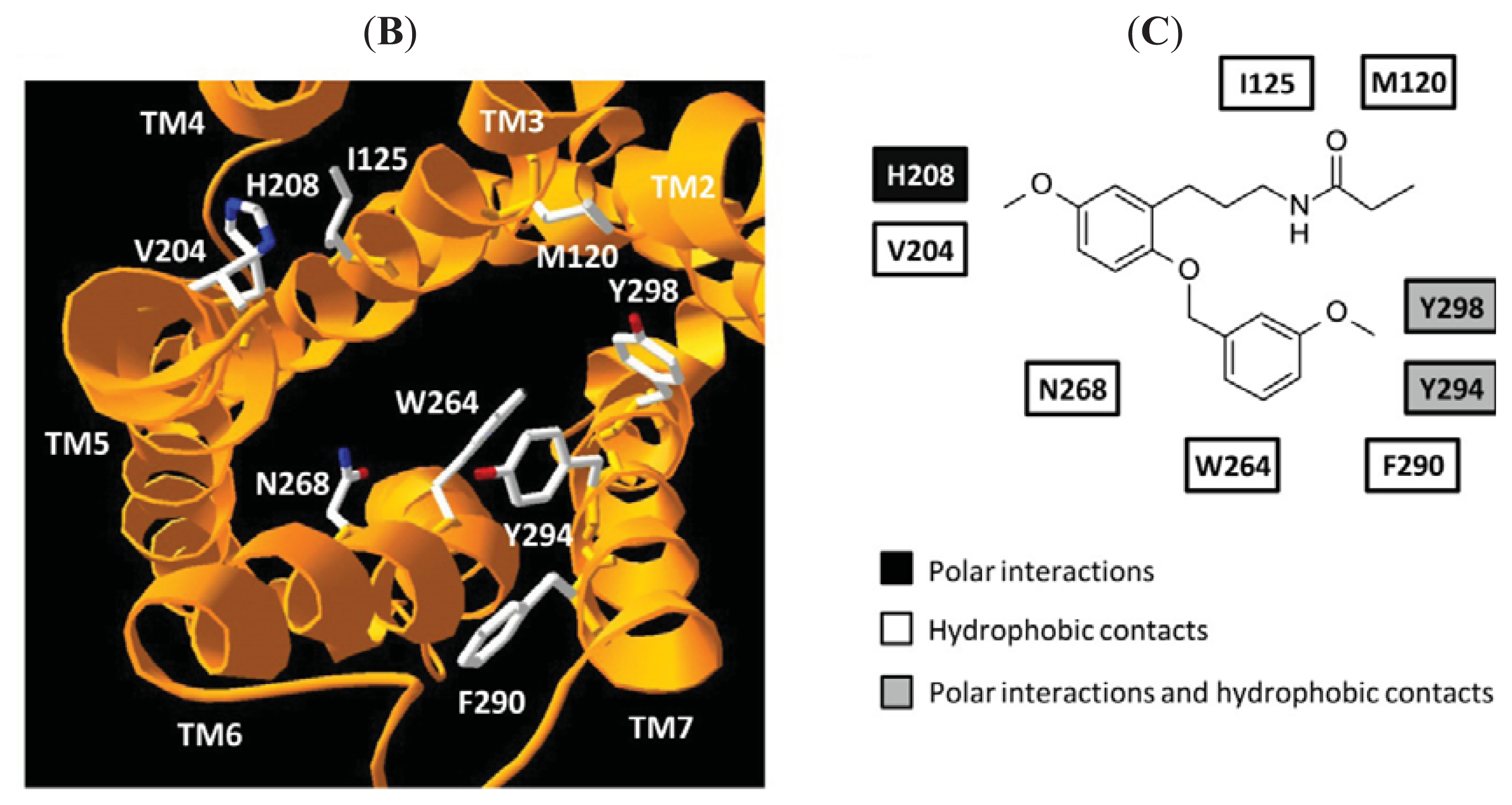
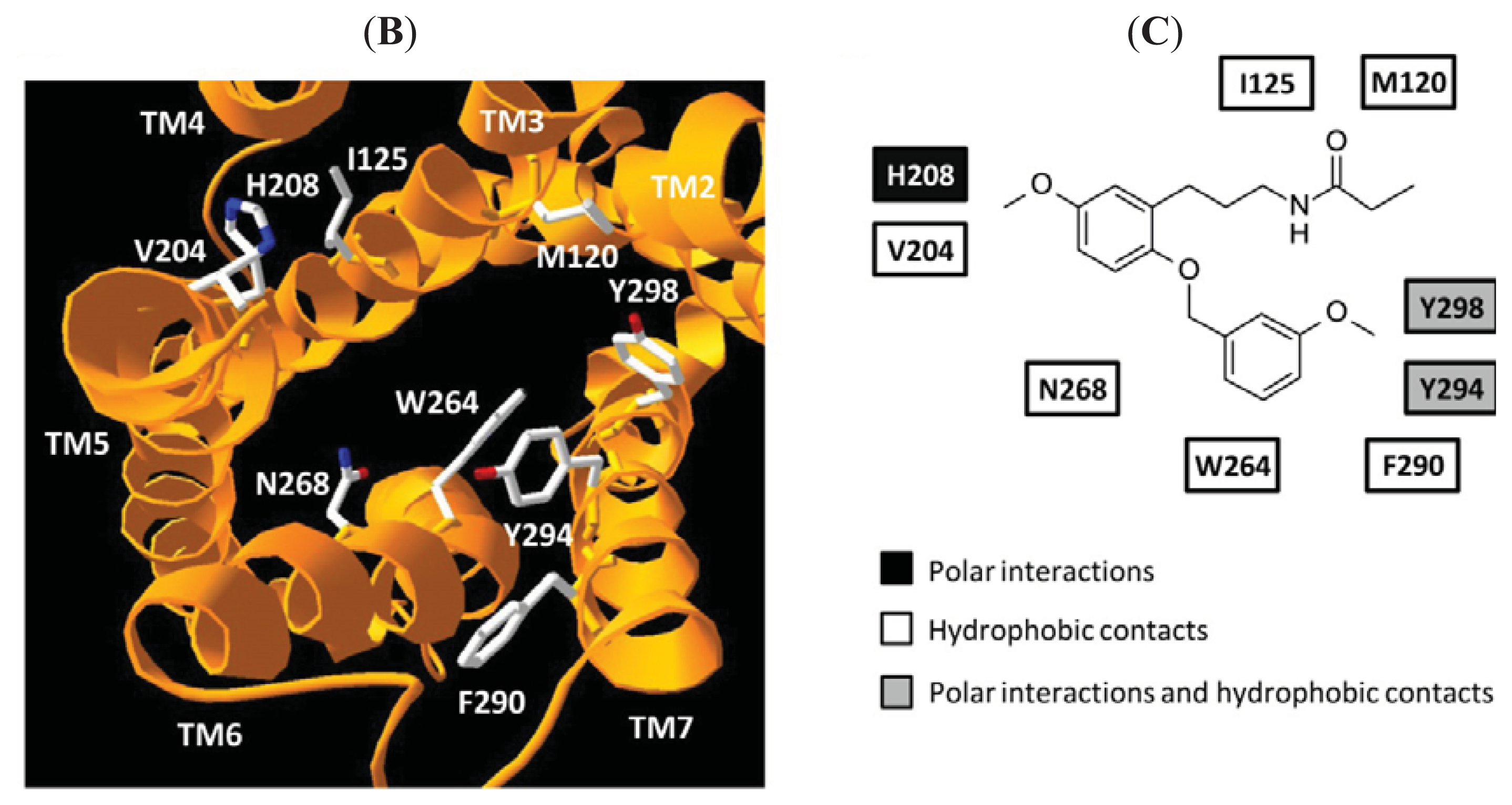
4. Homology Models of Melatonin Receptors
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Poeggeler, B.; Reiter, R.J.; Tan, D.X.; Chen, L.D.; Manchester, L.C. Melatonin, hydroxyl radical-mediated oxidative damage, and aging: A hypothesis. J. Pineal Res 1993, 14, 151–168. [Google Scholar]
- Kennaway, D.; Wright, H. Melatonin and circadian rhythms. Curr. Top. Med. Chem 2002, 2, 199–209. [Google Scholar]
- Geoffriau, M.; Claustrat, B.; Veldhuis, J. Estimation of frequently sampled nocturnal melatonin production in humans by deconvolution analysis: Evidence for episodic or ultradian secretion. J. Pineal Res 1999, 27, 139–144. [Google Scholar]
- Pandi-Perumal, S.; Srinivasan, V.; Maestroni, G.; Cardinali, D.; Poeggeler, B.; Hardeland, R. Melatonin: Nature’s most versatile biological signal? FEBS J 2006, 273, 2813–2838. [Google Scholar]
- Cardinali, D.; Furio, A.; Reyes, M.; Brusco, L. The use of chronobiotics in the resynchronization of the sleep-wake cycle. Cancer Causes Control 2006, 17, 601–609. [Google Scholar]
- Dawson, D.; Armstrong, S. Chronobiotics—Drugs that shift rhythms. Pharmacol. Ther 1996, 69, 15–36. [Google Scholar]
- Arendt, J.; Skene, D. Melatonin as a chronobiotic. Sleep Med. Rev 2005, 9, 25–39. [Google Scholar]
- Touitou, Y.; Bogdan, A. Promoting adjustment of the sleep-wake cycle by chronobiotics. Physiol. Behav 2007, 90, 294–300. [Google Scholar]
- Sack, R.L.; Auckley, D.; Auger, R.R.; Carskadon, M.A.; Wright, K.P.; Vitiello, M.V.; Zhdanova, I.V.; Medicine, A.A.O.S. Circadian rhythm sleep disorders: part I, basic principles, shift work and jet lag disorders. An American Academy of Sleep Medicine review. Sleep 2007, 30, 1460–1483. [Google Scholar]
- Kayumov, L.; Brown, G.; Jindal, R.; Buttoo, K.; Shapiro, C.M. A randomized, double-blind, placebo-controlled crossover study of the effect of exogenous melatonin on delayed sleep phase syndrome. Psychosom. Med 2001, 63, 40–48. [Google Scholar]
- Sack, R.L.; Auckley, D.; Auger, R.R.; Carskadon, M.A.; Wright, K.P.; Vitiello, M.V.; Zhdanova, I.V.; Medicine, A.A.O.S. Circadian rhythm sleep disorders: part II, advanced sleep phase disorder, delayed sleep phase disorder, free-running disorder, and irregular sleep-wake rhythm. An American Academy of Sleep Medicine review. Sleep 2007, 30, 1484–1501. [Google Scholar]
- Skene, D.J.; Arendt, J. Circadian rhythm sleep disorders in the blind and their treatment with melatonin. Sleep Med 2007, 8, 651–655. [Google Scholar]
- Herxheimer, A.; Petrie, K.J. Melatonin for the prevention and treatment of jet lag. Cochrane Database Syst. Rev. 2002. [Google Scholar] [CrossRef]
- Arendt, J. Melatonin and human rhythms. Chronobiol. Int 2006, 23, 21–37. [Google Scholar]
- Ekmekcioglu, C. Melatonin receptors in humans: biological role and clinical relevance. Biomed. Pharmacother 2006, 60, 97–108. [Google Scholar]
- Ram, P.; Dai, J.; Yuan, L.; Dong, C.; Kiefer, T.; Lai, L.; Hill, S. Involvement of the mt1 melatonin receptor in human breast cancer. Cancer Lett 2002, 179, 141–150. [Google Scholar]
- Carrillo-Vico, A.; Reiter, R.; Lardone, P.; Herrera, J.; Fernández-Montesinos, R.; Guerrero, J.; Pozo, D. The modulatory role of melatonin on immune responsiveness. Curr. Opin. Investig. Drugs 2006, 7, 423–431. [Google Scholar]
- Bondy, S.C.; Sharman, E.H. Melatonin and the aging brain. Neurochem. Int 2007, 50, 571–580. [Google Scholar]
- De Jonghe, A.; Korevaar, J.C.; van Munster, B.C.; de Rooij, S.E. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int. J. Geriatr. Psychiatry 2010, 25, 1201–1208. [Google Scholar]
- Jan, J.E.; Reiter, R.J.; Wong, P.K.; Bax, M.C.; Ribary, U.; Wasdell, M.B. Melatonin has membrane receptor-independent hypnotic action on neurons: An hypothesis. J. Pineal Res 2011, 50, 233–240. [Google Scholar]
- Larson, E.B.; Zollman, F.S. The effect of sleep medications on cognitive recovery from traumatic brain injury. J. Head Trauma Rehabil 2010, 25, 61–67. [Google Scholar]
- Reiter, R.J.; Tan, D.X.; Manchester, L.C.; Tamura, H. Melatonin defeats neurally-derived free radicals and reduces the associated neuromorphological and neurobehavioral damage. J. Physiol. Pharmacol 2007, 58, 5–22. [Google Scholar]
- Ebisawa, T.; Karne, S.; Lerner, M.; Reppert, S. Expression cloning of a high-affinity melatonin receptor from Xenopus dermal melanophores. Proc. Natl. Acad. Sci. USA 1994, 91, 6133–6137. [Google Scholar]
- Reppert, S.; Godson, C.; Mahle, C.; Weaver, D.; Slaugenhaupt, S.; Gusella, J. Molecular characterization of a second melatonin receptor expressed in human retina and brain: The Mel1b melatonin receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 8734–8738. [Google Scholar]
- Reppert, S.; Weaver, D.; Ebisawa, T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron 1994, 13, 1177–1185. [Google Scholar]
- Dubocovich, M.; Markowska, M. Functional MT1 and MT2 melatonin receptors in mammals. Endocrine 2005, 27, 101–110. [Google Scholar]
- Slaugenhaupt, S.; Roca, A.; Liebert, C.; Altherr, M.; Gusella, J.; Reppert, S. Mapping of the gene for the Mel1a-melatonin receptor to human chromosome 4 (MTNR1A) and mouse chromosome 8 (Mtnr1a). Genomics 1995, 27, 355–357. [Google Scholar]
- Reppert, S.; Weaver, D.; Ebisawa, T.; Mahle, C.; Kolakowski, L.J. Cloning of a melatonin-related receptor from human pituitary. FEBS Lett 1996, 386, 219–224. [Google Scholar]
- Levoye, A.; Dam, J.; Ayoub, M.; Guillaume, J.; Couturier, C.; Delagrange, P.; Jockers, R. The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J 2006, 25, 3012–3023. [Google Scholar]
- Becker-André, M.; Wiesenberg, I.; Schaeren-Wiemers, N.; André, E.; Missbach, M.; Saurat, J.; Carlberg, C. Pineal gland hormone melatonin binds and activates an orphan of the nuclear receptor superfamily. J. Biol. Chem 1994, 269, 28531–28534. [Google Scholar]
- Skwarlo-Sonta, K.; Majewski, P.; Markowska, M.; Oblap, R.; Olszanska, B. Bidirectional communication between the pineal gland and the immune system. Can. J. Physiol. Pharmacol 2003, 81, 342–349. [Google Scholar]
- Pandi-Perumal, S.; Trakht, I.; Srinivasan, V.; Spence, D.; Maestroni, G.; Zisapel, N.; Cardinali, D. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol 2008, 85, 335–353. [Google Scholar]
- Liu, C.; Weaver, D.; Jin, X.; Shearman, L.; Pieschl, R.; Gribkoff, V.; Reppert, S. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron 1997, 19, 91–102. [Google Scholar]
- Reiter, R. Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr. Rev 1991, 12, 151–180. [Google Scholar]
- Dubocovich, M.; Rivera-Bermudez, M.; Gerdin, M.; Masana, M. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front. Biosci 2003, 8, d1093–d1108. [Google Scholar]
- Clemens, J.; Jarzynka, M.; Witt-Enderby, P. Down-regulation of mt1 melatonin receptors in rat ovary following estrogen exposure. Life Sci 2001, 69, 27–35. [Google Scholar]
- Frungieri, M.; Mayerhofer, A.; Zitta, K.; Pignataro, O.; Calandra, R.; Gonzalez-Calvar, S. Direct effect of melatonin on Syrian hamster testes: Melatonin subtype 1a receptors, inhibition of androgen production, and interaction with the local corticotropin-releasing hormone system. Endocrinology 2005, 146, 1541–1552. [Google Scholar]
- Masana, M.; Doolen, S.; Ersahin, C.; Al-Ghoul, W.; Duckles, S.; Dubocovich, M.; Krause, D. MT(2) melatonin receptors are present and functional in rat caudal artery. J. Pharmacol. Exp. Ther 2002, 302, 1295–1302. [Google Scholar]
- Naji, L.; Carrillo-Vico, A.; Guerrero, J.; Calvo, J. Expression of membrane and nuclear melatonin receptors in mouse peripheral organs. Life Sci 2004, 74, 2227–2236. [Google Scholar]
- Zalatan, F.; Krause, J.; Blask, D. Inhibition of isoproterenol-induced lipolysis in rat inguinal adipocytes in vitro by physiological melatonin via a receptor-mediated mechanism. Endocrinology 2001, 142, 3783–3790. [Google Scholar]
- Guerrero, J.; Reiter, R. Melatonin-immune system relationships. Curr. Top. Med. Chem 2002, 2, 167–179. [Google Scholar]
- Brydon, L.; Roka, F.; Petit, L.; de Coppet, P.; Tissot, M.; Barrett, P.; Morgan, P.; Nanoff, C.; Strosberg, A.; Jockers, R. Dual signaling of human Mel1a melatonin receptors via G(i2), G(i3), and G(q/11) proteins. Mol. Endocrinol 1999, 13, 2025–2038. [Google Scholar]
- Masana, M.I.; Dubocovich, M.L. Melatonin receptor signaling: finding the path through the dark. Sci. STKE 2001, 2001. [Google Scholar] [CrossRef]
- Travnickova-Bendova, Z.; Cermakian, N.; Reppert, S.M.; Sassone-Corsi, P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl. Acad. Sci. USA 2002, 99, 7728–7733. [Google Scholar]
- Jin, X.; von Gall, C.; Pieschl, R.; Gribkoff, V.; Stehle, J.; Reppert, S.; Weaver, D. Targeted disruption of the mouse Mel(1b) melatonin receptor. Mol. Cell. Biol 2003, 23, 1054–1060. [Google Scholar]
- Chan, A.; Lai, F.; Lo, R.; Voyno-Yasenetskaya, T.; Stanbridge, E.; Wong, Y. Melatonin mt1 and MT2 receptors stimulate c-Jun N-terminal kinase via pertussis toxin-sensitive and -insensitive G proteins. Cell Signal 2002, 14, 249–257. [Google Scholar]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar]
- van Dam, H.; Duyndam, M.; Rottier, R.; Bosch, A.; de Vries-Smits, L.; Herrlich, P.; Zantema, A.; Angel, P.; van der Eb, A.J. Heterodimer formation of cJun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J 1993, 12, 479–487. [Google Scholar]
- Zinck, R.; Cahill, M.A.; Kracht, M.; Sachsenmaier, C.; Hipskind, R.A.; Nordheim, A. Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress-activated protein kinase pathways that converge on Elk-1. Mol. Cell. Biol 1995, 15, 4930–4938. [Google Scholar]
- Garcia-Mauriño, S.; Gonzalez-Haba, M.G.; Calvo, J.R.; Rafii-El-Idrissi, M.; Sanchez-Margalet, V.; Goberna, R.; Guerrero, J.M. Melatonin enhances IL-2, IL-6, and IFN-gamma production by human circulating CD4+ cells: A possible nuclear receptor-mediated mechanism involving T helper type 1 lymphocytes and monocytes. J. Immunol 1997, 159, 574–581. [Google Scholar]
- Lau, W.W.; Ng, J.K.; Lee, M.M.; Chan, A.S.; Wong, Y.H. Interleukin-6 autocrine signaling mediates melatonin MT(1/2) receptor-induced STAT3 Tyr(705) phosphorylation. J. Pineal Res 2012, 52, 477–489. [Google Scholar]
- Eddahri, F.; Denanglaire, S.; Bureau, F.; Spolski, R.; Leonard, W.J.; Leo, O.; Andris, F. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood 2009, 113, 2426–2433. [Google Scholar]
- Shen, L.; Fahey, J.V.; Hussey, S.B.; Asin, S.N.; Wira, C.R.; Fanger, M.W. Synergy between IL-8 and GM-CSF in reproductive tract epithelial cell secretions promotes enhanced neutrophil chemotaxis. Cell. Immunol 2004, 230, 23–32. [Google Scholar]
- Drazen, D.; Nelson, R. Melatonin receptor subtype MT2 (Mel 1b) and not mt1 (Mel 1a) is associated with melatonin-induced enhancement of cell-mediated and humoral immunity. Neuroendocrinology 2001, 74, 178–184. [Google Scholar]
- Drazen, D.; Bilu, D.; Bilbo, S.; Nelson, R. Melatonin enhancement of splenocyte proliferation is attenuated by luzindole, a melatonin receptor antagonist. Am. J. Physiol. Regul. Integr. Comp. Physiol 2001, 280, R1476–R1482. [Google Scholar]
- Weil, Z.; Hotchkiss, A.; Gatien, M.; Pieke-Dahl, S.; Nelson, R. Melatonin receptor (MT1) knockout mice display depression-like behaviors and deficits in sensorimotor gating. Brain Res. Bull 2006, 68, 425–429. [Google Scholar]
- Larson, J.; Jessen, R.; Uz, T.; Arslan, A.; Kurtuncu, M.; Imbesi, M.; Manev, H. Impaired hippocampal long-term potentiation in melatonin MT2 receptor-deficient mice. Neurosci. Lett 2006, 393, 23–26. [Google Scholar]
- Von Gall, C.; Weaver, D.; Moek, J.; Jilg, A.; Stehle, J.; Korf, H. Melatonin plays a crucial role in the regulation of rhythmic clock gene expression in the mouse pars tuberalis. Ann. N Y Acad. Sci 2005, 1040, 508–511. [Google Scholar]
- Dubocovich, M.; Hudson, R.; Sumaya, I.; Masana, M.; Manna, E. Effect of MT1 melatonin receptor deletion on melatonin-mediated phase shift of circadian rhythms in the C57BL/6 mouse. J. Pineal Res 2005, 39, 113–120. [Google Scholar]
- Gerdin, M.; Masana, M.; Rivera-Bermúdez, M.; Hudson, R.; Earnest, D.; Gillette, M.; Dubocovich, M. Melatonin desensitizes endogenous MT2 melatonin receptors in the rat suprachiasmatic nucleus: Relevance for defining the periods of sensitivity of the mammalian circadian clock to melatonin. FASEB J 2004, 18, 1646–1656. [Google Scholar]
- Uchikawa, O.; Fukatsu, K.; Tokunoh, R.; Kawada, M.; Matsumoto, K.; Imai, Y.; Hinuma, S.; Kato, K.; Nishikawa, H.; Hirai, K.; et al. Synthesis of a novel series of tricyclic indan derivatives as melatonin receptor agonists. J. Med. Chem 2002, 45, 4222–4239. [Google Scholar]
- Dubocovich, M.; Takahashi, J. Use of 2-[125I]iodomelatonin to characterize melatonin binding sites in chicken retina. Proc. Natl. Acad. Sci. USA 1987, 84, 3916–3920. [Google Scholar]
- Nonno, R.; Lucini, V.; Pannacci, M.; Mazzucchelli, C.; Angeloni, D.; Fraschini, F.; Stankov, B. Pharmacological characterization of the human melatonin Mel1a receptor following stable transfection into NIH3T3 cells. Br. J. Pharmacol 1998, 124, 485–492. [Google Scholar]
- Mulchahey, J.; Goldwater, D.; Zemlan, F. A single blind, placebo controlled, across groups dose escalation study of the safety, tolerability, pharmacokinetics and pharmacodynamics of the melatonin analog beta-methyl-6-chloromelatonin. Life Sci 2004, 75, 1843–1856. [Google Scholar]
- Hardeland, R. Investigational melatonin receptor agonists. Expert Opin. Investig. Drugs 2010, 19, 747–764. [Google Scholar]
- Zlotos, D.P. Recent advances in melatonin receptor ligands. Arch. Pharm. (Weinheim) 2005, 338, 229–247. [Google Scholar]
- Mor, M.; Rivara, S.; Pala, D.; Bedini, A.; Spadoni, G.; Tarzia, G. Recent advances in the development of melatonin MT(1) and MT(2) receptor agonists. Expert Opin. Ther. Pat 2010, 20, 1059–1077. [Google Scholar]
- Zlotos, D.P. Recent progress in the development of agonists and antagonists for melatonin receptors. Curr. Med. Chem 2012, 19, 3532–3549. [Google Scholar]
- Kato, K.; Hirai, K.; Nishiyama, K.; Uchikawa, O.; Fukatsu, K.; Ohkawa, S.; Kawamata, Y.; Hinuma, S.; Miyamoto, M. Neurochemical properties of ramelteon (TAK-375), a selective MT1/MT2 receptor agonist. Neuropharmacology 2005, 48, 301–310. [Google Scholar]
- Bourin, M.; Mocaër, E.; Porsolt, R. Antidepressant-like activity of S 20098 (agomelatine) in the forced swimming test in rodents: involvement of melatonin and serotonin receptors. J. Psychiatry Neurosci 2004, 29, 126–133. [Google Scholar]
- Millan, M.; Gobert, A.; Lejeune, F.; Dekeyne, A.; Newman-Tancredi, A.; Pasteau, V.; Rivet, J.; Cussac, D. The novel melatonin agonist agomelatine (S20098) is an antagonist at 5-hydroxytryptamine2C receptors, blockade of which enhances the activity of frontocortical dopaminergic and adrenergic pathways. J. Pharmacol. Exp. Ther 2003, 306, 954–964. [Google Scholar]
- Audinot, V.; Mailliet, F.; Lahaye-Brasseur, C.; Bonnaud, A.; Le Gall, A.; Amossé, C.; Dromaint, S.; Rodriguez, M.; Nagel, N.; Galizzi, J.; et al. New selective ligands of human cloned melatonin MT1 and MT2 receptors. Naunyn. Schmiedeberg Arch. Pharmacol 2003, 367, 553–561. [Google Scholar]
- Ettaoussi, M.; Sabaouni, A.; Rami, M.; Boutin, J.A.; Delagrange, P.; Renard, P.; Spedding, M.; Caignard, D.H.; Berthelot, P.; Yous, S. Design, synthesis and pharmacological evaluation of new series of naphthalenic analogues as melatoninergic (MT1/MT2) and serotoninergic 5-HT2C dual ligands (I). Eur. J. Med. Chem 2012, 49, 310–323. [Google Scholar]
- Hardeland, R. Tasimelteon, a melatonin agonist for the treatment of insomnia and circadian rhythm sleep disorders. Curr. Opin. Investig. Drugs 2009, 10, 691–701. [Google Scholar]
- Dubocovich, M.; Masana, M.; Iacob, S.; Sauri, D. Melatonin receptor antagonists that differentiate between the human Mel1a and Mel1b recombinant subtypes are used to assess the pharmacological profile of the rabbit retina ML1 presynaptic heteroreceptor. Naunyn. Schmiedeberg Arch. Pharmacol 1997, 355, 365–375. [Google Scholar]
- Teh, M.; Sugden, D. Comparison of the structure-activity relationships of melatonin receptor agonists and antagonists: Lengthening the N-acyl side-chain has differing effects on potency on Xenopus melanophores. Naunyn. Schmiedeberg Arch. Pharmacol 1998, 358, 522–528. [Google Scholar]
- Faust, R.; Garratt, P.J.; Jones, R.; Yeh, L.K.; Tsotinis, A.; Panoussopoulou, M.; Calogeropoulou, T.; Teh, M.T.; Sugden, D. Mapping the melatonin receptor. 6. Melatonin agonists and antagonists derived from 6H-isoindolo[2,1-a]indoles, 5,6-dihydroindolo[2,1-a]isoquinolines, and 6,7-dihydro-5H-benzo[c]azepino[2,1-a]indoles. J. Med. Chem 2000, 43, 1050–1061. [Google Scholar]
- Wallez, V.; Durieux-Poissonnier, S.; Chavatte, P.; Boutin, J.; Audinot, V.; Nicolas, J.; Bennejean, C.; Delagrange, P.; Renard, P.; Lesieur, D. Synthesis and structure-affinity-activity relationships of novel benzofuran derivatives as MT(2) melatonin receptor selective ligands. J. Med. Chem 2002, 45, 2788–2800. [Google Scholar]
- Descamps-François, C.; Yous, S.; Chavatte, P.; Audinot, V.; Bonnaud, A.; Boutin, J.; Delagrange, P.; Bennejean, C.; Renard, P.; Lesieur, D. Design and synthesis of naphthalenic dimers as selective MT1 melatoninergic ligands. J. Med. Chem 2003, 46, 1127–1129. [Google Scholar]
- Rivara, S.; Mor, M.; Bedini, A.; Spadoni, G.; Tarzia, G. Melatonin receptor agonists: SAR and applications to the treatment of sleep-wake disorders. Curr. Top. Med. Chem 2008, 8, 954–968. [Google Scholar]
- Rivara, S.; Lodola, A.; Mor, M.; Bedini, A.; Spadoni, G.; Lucini, V.; Pannacci, M.; Fraschini, F.; Scaglione, F.; Sanchez, R.O.; et al. N-(substituted-anilinoethyl)amides: design, synthesis, and pharmacological characterization of a new class of melatonin receptor ligands. J. Med. Chem 2007, 50, 6618–6626. [Google Scholar]
- Rivara, S.; Pala, D.; Lodola, A.; Mor, M.; Lucini, V.; Dugnani, S.; Scaglione, F.; Bedini, A.; Lucarini, S.; Tarzia, G.; et al. MT(1)-Selective Melatonin Receptor Ligands: Synthesis, Pharmacological Evaluation, and Molecular Dynamics Investigation of N-{[(3-O-Substituted) anilino]alkyl}amides. ChemMedChem 2012, 7, 1954–1964. [Google Scholar]
- Sugden, D.; Pickering, H.; Teh, M.T.; Garratt, P.J. Melatonin receptor pharmacology: toward subtype specificity. Biol. Cell 1997, 89, 531–537. [Google Scholar]
- Navajas, C.; Kokkola, T.; Poso, A.; Honka, N.; Gynther, J.; Laitinen, J.T. A rhodopsin-based model for melatonin recognition at its G protein-coupled receptor. Eur. J. Pharmacol 1996, 304, 173–183. [Google Scholar]
- Garratt, P.; Travard, S.; Vonhoff, S.; Tsotinis, A.; Sugden, D. Mapping the melatonin receptor. 4. Comparison of the binding affinities of a series of substituted phenylalkyl amides. J. Med. Chem 1996, 39, 1797–1805. [Google Scholar]
- Spadoni, G.; Mor, M.; Tarzia, G. Structure-affinity relationships of indole-based melatonin analogs. Biol. Signals Recept 1999, 8, 15–23. [Google Scholar]
- Mor, M.; Rivara, S.; Silva, C.; Bordi, F.; Plazzi, P.V.; Spadoni, G.; Diamantini, G.; Balsamini, C.; Tarzia, G.; Fraschini, F.; et al. Melatonin receptor ligands: synthesis of new melatonin derivatives and comprehensive comparative molecular field analysis (CoMFA) study. J. Med. Chem 1998, 41, 3831–3844. [Google Scholar]
- Sugden, D.; Chong, N.; Lewis, D. Structural requirements at the melatonin receptor. Br. J. Pharmacol 1995, 114, 618–623. [Google Scholar]
- Spadoni, G.; Stankov, B.; Duranti, A.; Biella, G.; Lucini, V.; Salvatori, A.; Fraschini, F. 2-Substituted 5-methoxy-N-acyltryptamines: synthesis, binding affinity for the melatonin receptor, and evaluation of the biological activity. J. Med. Chem 1993, 36, 4069–4074. [Google Scholar]
- Beresford, I.; Browning, C.; Starkey, S.; Brown, J.; Foord, S.; Coughlan, J.; North, P.; Dubocovich, M.; Hagan, R. GR196429: A nonindolic agonist at high-affinity melatonin receptors. J. Pharmacol. Exp. Ther 1998, 285, 1239–1245. [Google Scholar]
- Mody, S.; Hu, Y.; Ho, M.; Wong, Y. In search of novel and therapeutically significant melatoninergic ligands. Recent Pat. CNS Drug Discov 2007, 2, 241–245. [Google Scholar]
- Hu, Y.; Ho, M.; Chan, K.; New, D.; Wong, Y. Synthesis of substituted N-[3-(3-methoxyphenyl)propyl] amides as highly potent MT(2)-selective melatonin ligands. Bioorg. Med. Chem. Lett 2010, 20, 2582–2585. [Google Scholar]
- Chan, K.H.; Hu, Y.; Ho, M.K.; Wong, Y.H. Characterization of Substituted Phenylpropylamides as Highly Selective Agonists at the Melatonin MT(2) Receptor. Curr. Med. Chem 2013, 20, 289–300. [Google Scholar]
- Gerdin, M.; Mseeh, F.; Dubocovich, M. Mutagenesis studies of the human MT2 melatonin receptor. Biochem. Pharmacol 2003, 66, 315–320. [Google Scholar]
- Kokkola, T.; Watson, M.A.; White, J.; Dowell, S.; Foord, S.M.; Laitinen, J.T. Mutagenesis of human Mel1a melatonin receptor expressed in yeast reveals domains important for receptor function. Biochem. Biophys. Res. Commun 1998, 249, 531–536. [Google Scholar]
- Conway, S.; Mowat, E.; Drew, J.; Barrett, P.; Delagrange, P.; Morgan, P. Serine residues 110 and 114 are required for agonist binding but not antagonist binding to the melatonin MT(1) receptor. Biochem. Biophys. Res. Commun 2001, 282, 1229–1236. [Google Scholar]
- Mazna, P.; Obsilova, V.; Jelinkova, I.; Balik, A.; Berka, K.; Sovova, Z.; Ettrich, R.; Svoboda, P.; Obsil, T.; Teisinger, J. Molecular modeling of human MT2 melatonin receptor: the role of Val204, Leu272 and Tyr298 in ligand binding. J. Neurochem 2004, 91, 836–842. [Google Scholar]
- Mazna, P.; Berka, K.; Jelinkova, I.; Balik, A.; Svoboda, P.; Obsilova, V.; Obsil, T.; Teisinger, J. Ligand binding to the human MT2 melatonin receptor: the role of residues in transmembrane domains 3, 6, and 7. BioChem. Biophys Res. Commun 2005, 332, 726–734. [Google Scholar]
- Farce, A.; Chugunov, A.O.; Logé, C.; Sabaouni, A.; Yous, S.; Dilly, S.; Renault, N.; Vergoten, G.; Efremov, R.G.; Lesieur, D.; et al. Homology modeling of MT1 and MT2 receptors. Eur. J. Med. Chem 2008, 43, 1926–1944. [Google Scholar]
- Mor, M.; Spadoni, G.; Di Giacomo, B.; Diamantini, G.; Bedini, A.; Tarzia, G.; Plazzi, P.V.; Rivara, S.; Nonno, R.; Lucini, V.; et al. Synthesis, pharmacological characterization and QSAR studies on 2-substituted indole melatonin receptor ligands. Bioorg. Med. Chem 2001, 9, 1045–1057. [Google Scholar]
- Rivara, S.; Lorenzi, S.; Mor, M.; Plazzi, P.V.; Spadoni, G.; Bedini, A.; Tarzia, G. Analysis of structure-activity relationships for MT2 selective antagonists by melatonin MT1 and MT2 receptor models. J. Med. Chem 2005, 48, 4049–4060. [Google Scholar]
- Shi, L.; Javitch, J.A. The binding site of aminergic G protein-coupled receptors: the transmembrane segments and second extracellular loop. Annu. Rev. Pharmacol. Toxicol 2002, 42, 437–467. [Google Scholar]
- Kristiansen, K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol. Ther 2004, 103, 21–80. [Google Scholar]
- Shi, L.; Liapakis, G.; Xu, R.; Guarnieri, F.; Ballesteros, J.A.; Javitch, J.A. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J. Biol. Chem 2002, 277, 40989–40996. [Google Scholar]
- Gloriam, D.E.; Foord, S.M.; Blaney, F.E.; Garland, S.L. Definition of the G protein-coupled receptor transmembrane bundle binding pocket and calculation of receptor similarities for drug design. J. Med. Chem 2009, 52, 4429–4442. [Google Scholar]
- Dubocovich, M.; Delagrange, P.; Krause, D.; Sugden, D.; Cardinali, D.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, Classification, and Pharmacology of G Protein-Coupled Melatonin Receptors. Pharmacol. Rev 2010, 62, 343–380. [Google Scholar]
- Renault, N.; Gohier, A.; Chavatte, P.; Farce, A. Novel structural insights for drug design of selective 5-HT(2C) inverse agonists from a ligand-biased receptor model. Eur. J. Med. Chem 2010, 45, 5086–5099. [Google Scholar]
- Farce, A.; Dilly, S.; Yous, S.; Berthelot, P.; Chavatte, P. Homology modelling of the serotoninergic 5-HT2c receptor. J. Enzyme Inhib. Med. Chem 2006, 21, 285–292. [Google Scholar]
- Pala, D.; Beuming, T.; Sherman, W.; Lodola, A.; Rivara, S.; Mor, M. Structure-based virtual screening of MT2 melatonin receptor: influence of template choice and structural refinement. J. Chem. Inf. Model 2013, 53, 821–835. [Google Scholar]
- Kufareva, I.; Rueda, M.; Katritch, V.; Stevens, R.C.; Abagyan, R.; participants, G.D. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 2011, 19, 1108–1126. [Google Scholar]
- Cherezov, V.; Rosenbaum, D.; Hanson, M.; Rasmussen, S.; Thian, F.; Kobilka, T.; Choi, H.; Kuhn, P.; Weis, W.; Kobilka, B.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | General TM numbering | Binding/functional characterizatics |
|---|---|---|
| S110A | 3.35 | Kd increased by 8-fold, Bmax reduced by 10-fold. Ki increased by 9-fold. No change in Ki of luzindole [96]. |
| S114A | 3.39 | Kd increased by 9-fold, Bmax reduced by 4-fold. Ki increased by 4-fold. No change in Ki of luzindole [96]. |
| H195A | 5.46 | EC50 of melatonin or 2-iodomelatonin reduced by 3–6 fold in yeast CPRG assay [95]. |
| Mutation | General TM numbering | Binding/functional characterizatics |
|---|---|---|
| M120A | 3.32 | No change in Kd, Bmax reduced by 3-fold. Ki increased by 2-fold. Ki of 4P-PDOT reduced by 6-fold [98]. |
| G121A | 3.33 | No change in Kd or Bmax. Ki increased by 3-fold [98]. |
| G121I | 3.33 | No change in Kd or Bmax. Ki increased by 2-fold [98]. |
| S123A | 3.35 | No change in Kd or Ki. Bmax reduced by 5-fold [94]. |
| S127A | 3.39 | No change in Kd or Ki. Bmax reduced by 3-fold [94]. |
| I125A | 3.37 | No change in Kd or Bmax. No change in Ki [98]. |
| N175A | 4.60 | No change in Kd or Bmax. Ki increased by 4-fold [94]. |
| V204A | 5.42 | NSB [97] |
| H208A | 5.46 | Kd increased by 2-fold, Ki increased by 4-fold. No change in Bmax [94]. |
| F257A | 6.41 | No change in Kd or Bmax. No change in Ki [94]. |
| W264A | 6.48 | Kd reduced by 2-fold, no change in Ki. Bmax reduced by 23-fold [94]. |
| N268A | 6.52 | NSB [98] |
| N268D | 6.52 | NSB [98] |
| N268L | 6.52 | NSB [98] |
| N268Q | 6.52 | No change in Kd or Bmax. No change in Ki. [98] |
| L272A | 6.56 | NSB [97] |
| A275I | 6.59 | NSB [98] |
| A275V | 6.59 | No change in Kd or Bmax. No change in Ki. [98] |
| V291A | 7.36 | NSB [98] |
| V291I | 7.36 | NSB [98] |
| L295A | 7.40 | NSB [98] |
| L295I | 7.40 | NSB [98] |
| L295V | 7.40 | NSB [98] |
| Y298A | 7.43 | NSB [97] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chan, K.H.; Wong, Y.H. A Molecular and Chemical Perspective in Defining Melatonin Receptor Subtype Selectivity. Int. J. Mol. Sci. 2013, 14, 18385-18406. https://doi.org/10.3390/ijms140918385
Chan KH, Wong YH. A Molecular and Chemical Perspective in Defining Melatonin Receptor Subtype Selectivity. International Journal of Molecular Sciences. 2013; 14(9):18385-18406. https://doi.org/10.3390/ijms140918385
Chicago/Turabian StyleChan, King Hang, and Yung Hou Wong. 2013. "A Molecular and Chemical Perspective in Defining Melatonin Receptor Subtype Selectivity" International Journal of Molecular Sciences 14, no. 9: 18385-18406. https://doi.org/10.3390/ijms140918385