Phenoxybenzamine Is Neuroprotective in a Rat Model of Severe Traumatic Brain Injury

Abstract
:1. Introduction
2. Results and Discussion
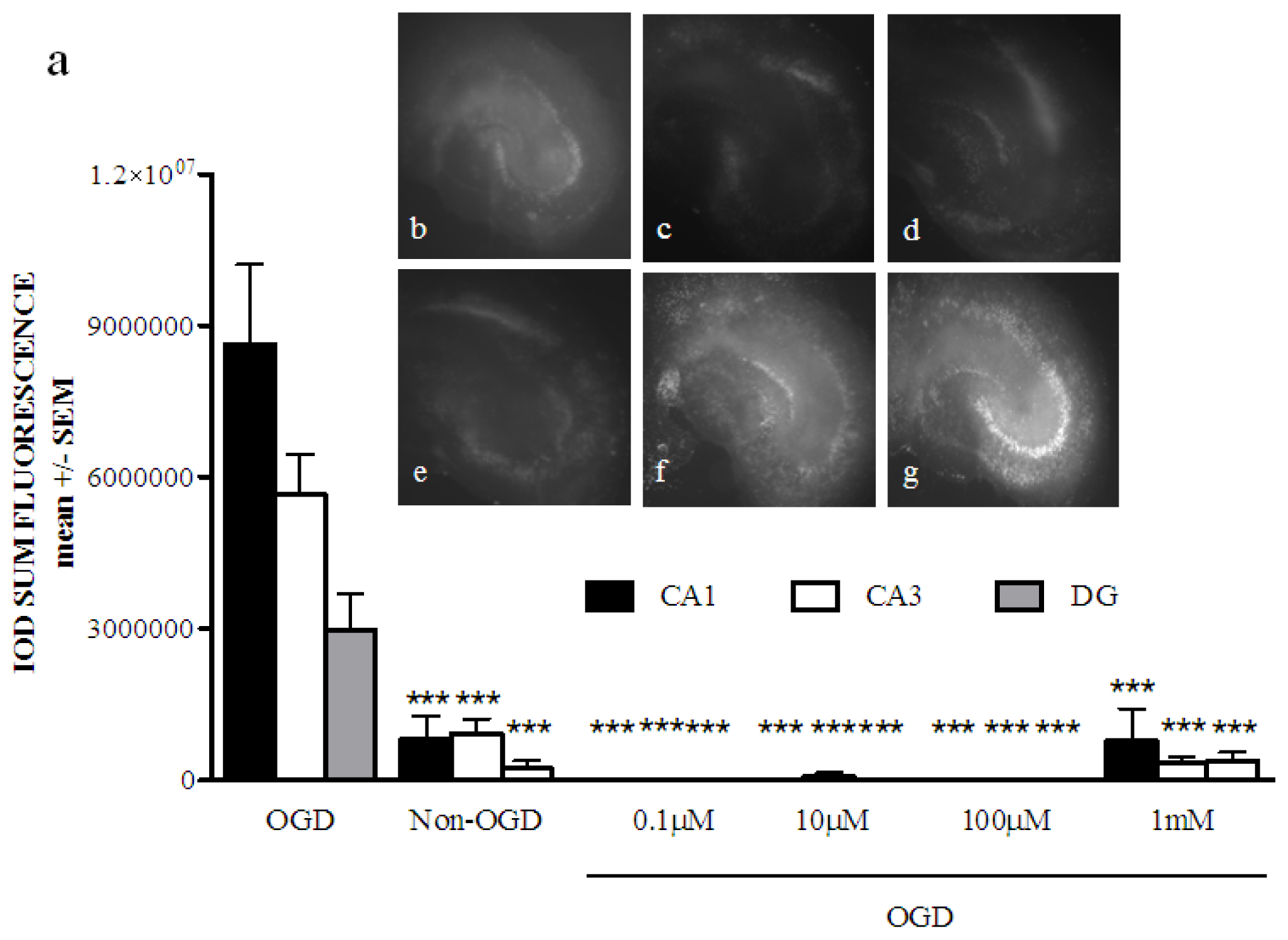
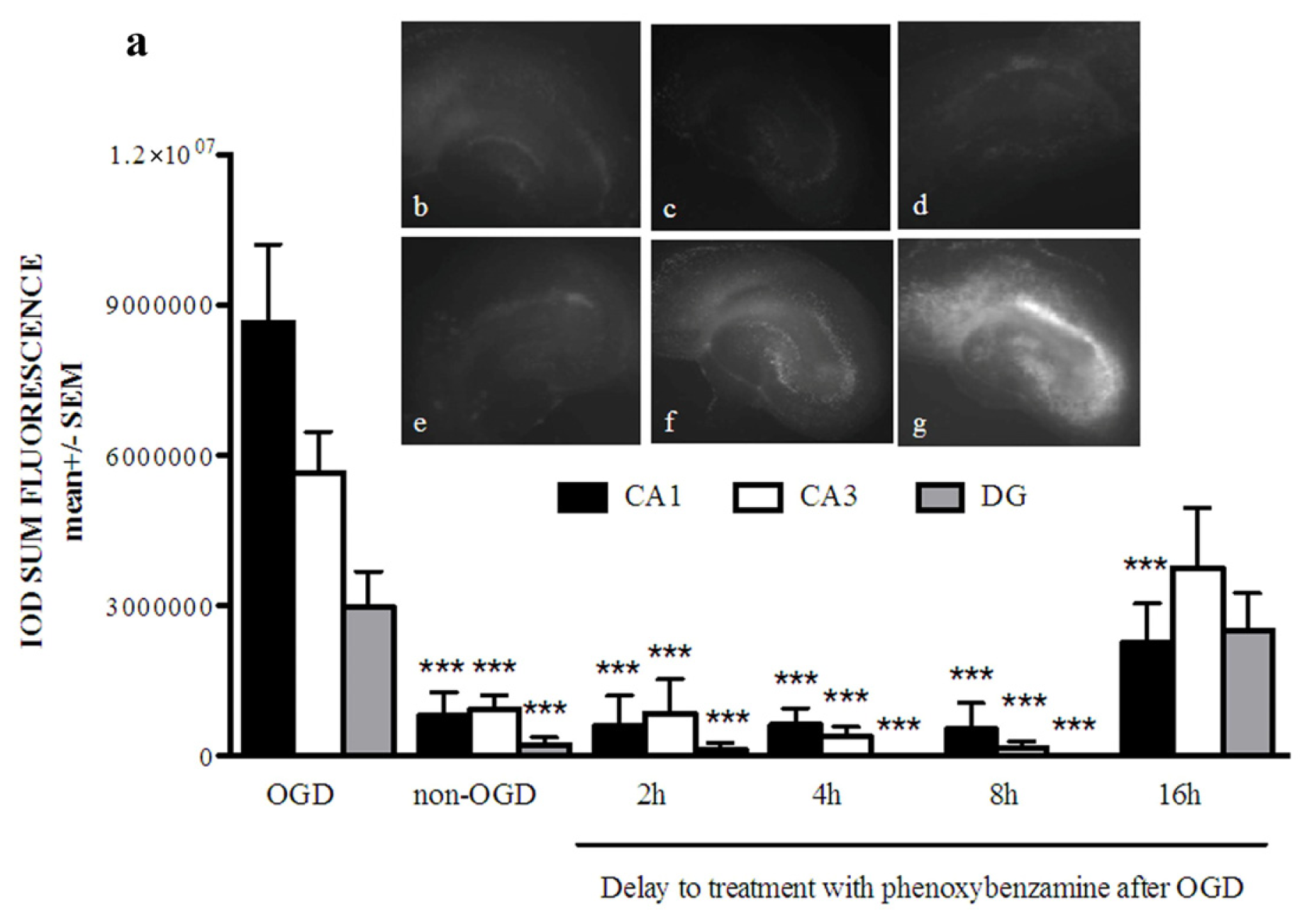
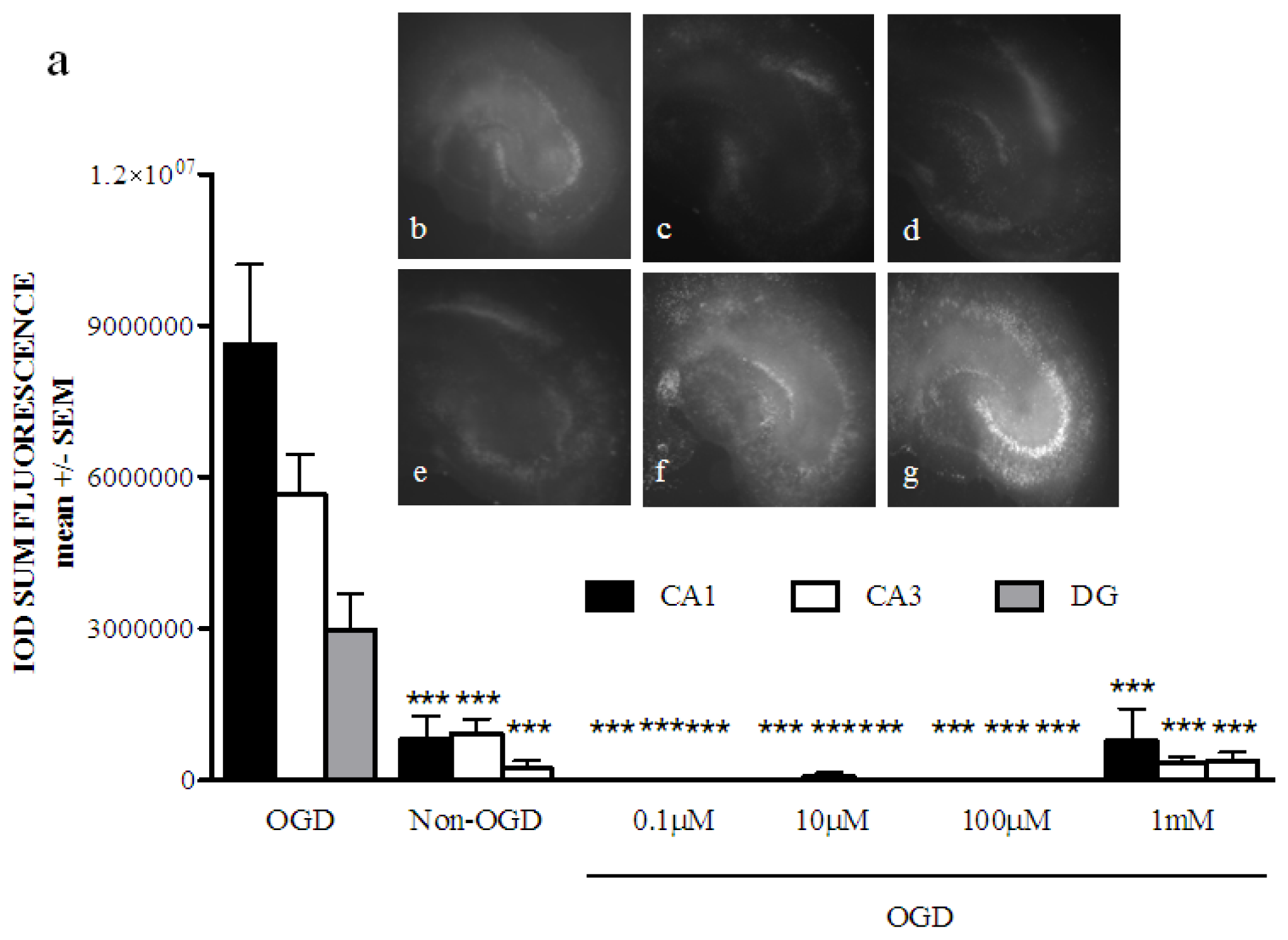
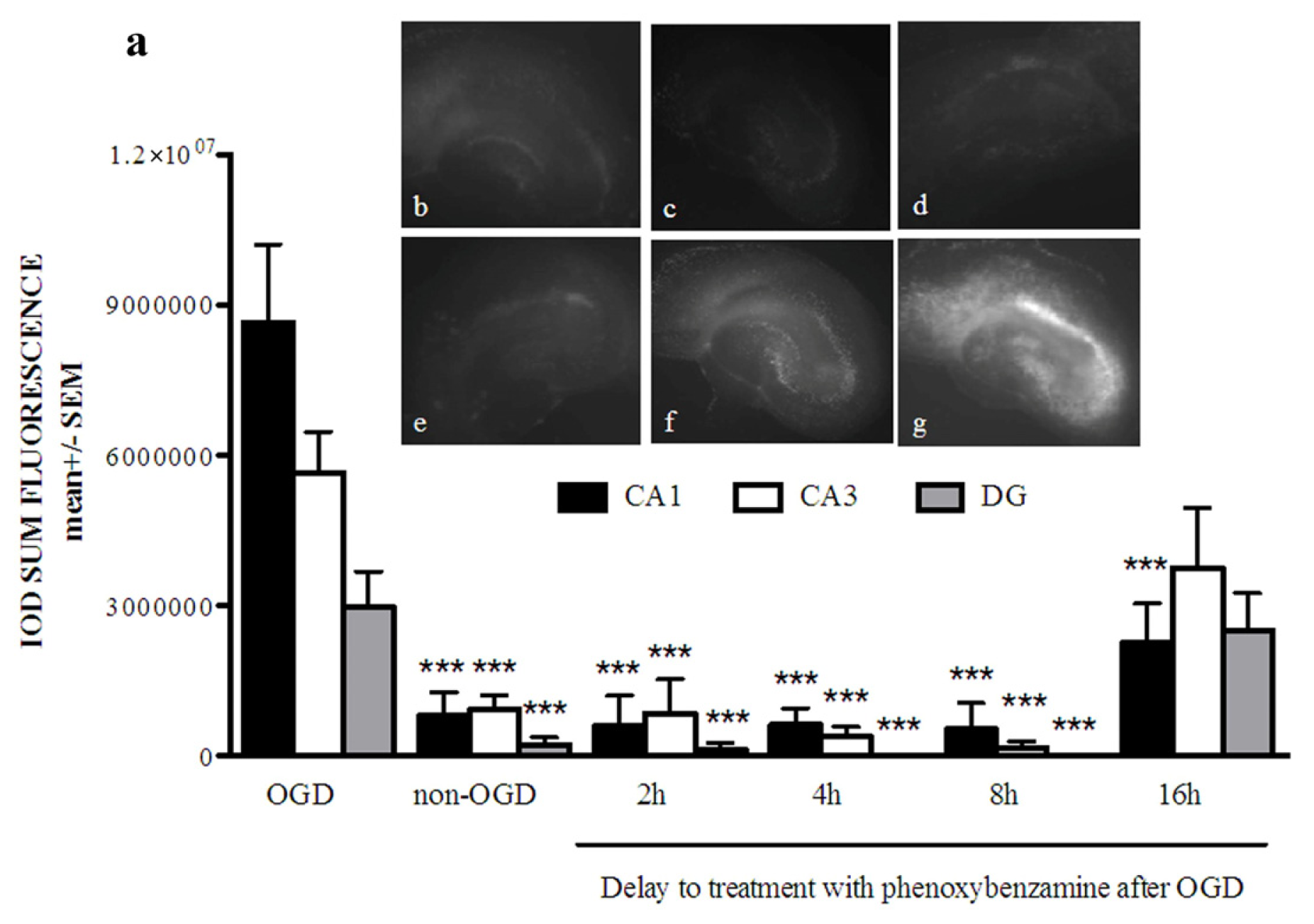
2.1. Phenoxybenzamine Prevents Hippocampal Cell Death after Oxygen Glucose Deprivation
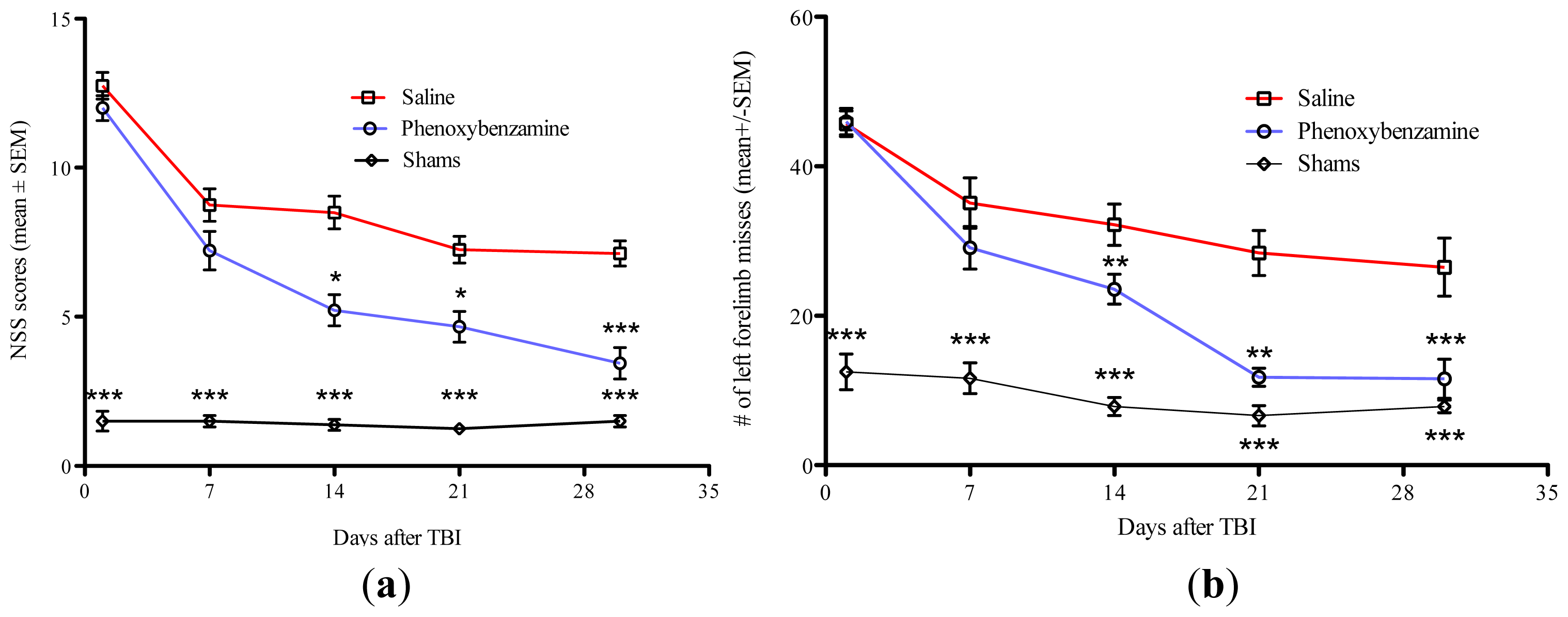
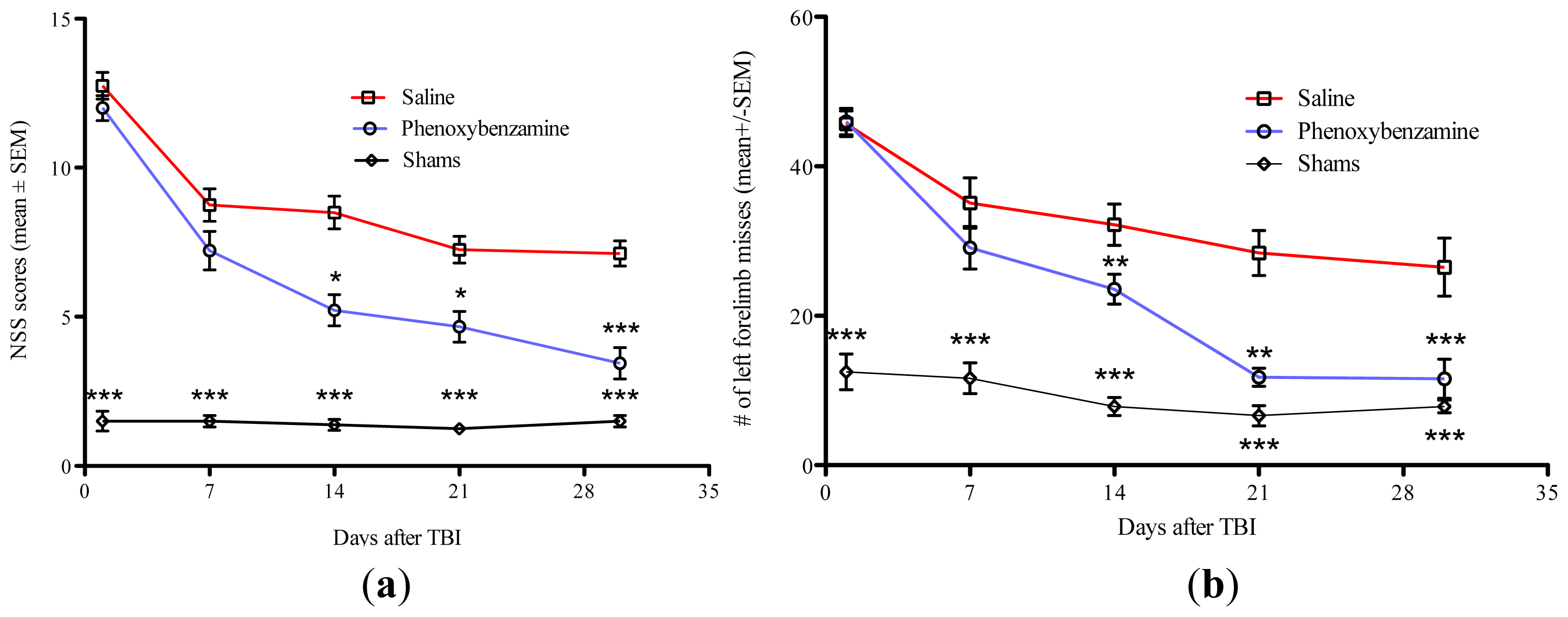
2.2. Phenoxybenzamine Prevents Neurological Dysfunction that Occurs as a Result of Severe TBI
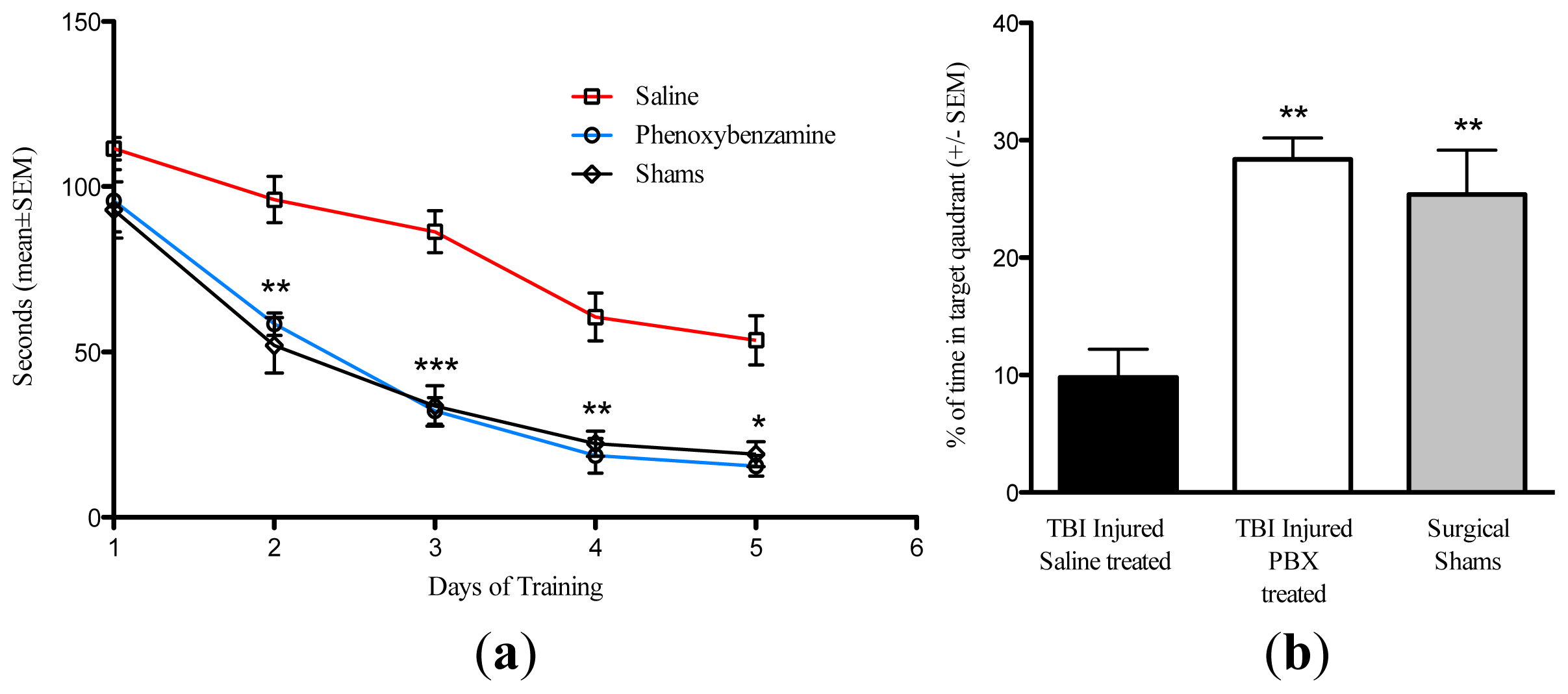
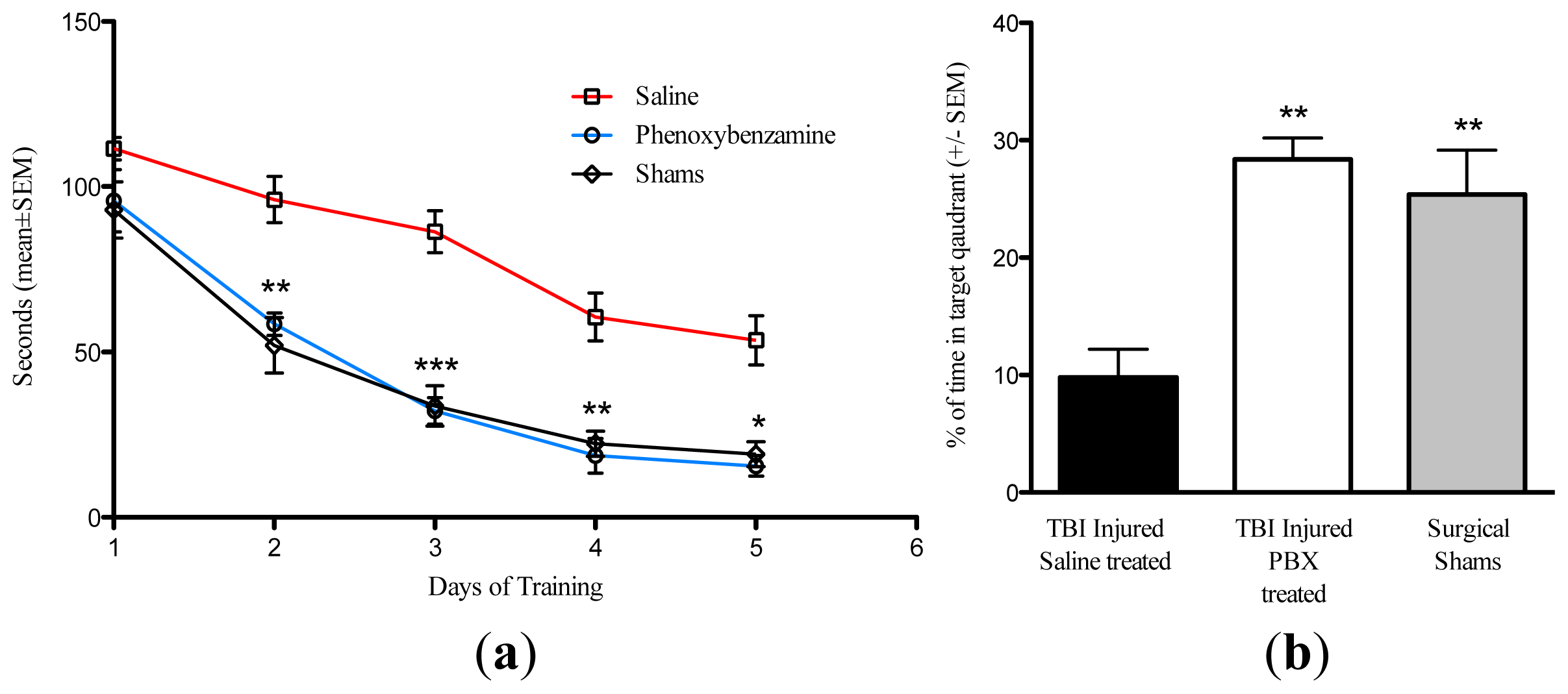
2.3. Phenoxybenzamine Reduces Cognitive Impairment Associated with Severe TBI
2.4. Phenoxybenzamine Reduces the Expression of Pro-Inflammatory Genes
2.5. Discussion
3. Experimental Section
3.1. Rat Organotypic Hippocampal Slice Cultures
3.2. TBI Procedure
3.3. Neurological Severity Scoring
3.4. Foot Faults Assessments
3.5. Assessment of Cognitive Function
3.6. RNA Isolation/Qrt-PCR
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Faul, M.; Xu, L.; Wald, M.; Coronado, V. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006; CDC: Atlanta, GA, USA, 2006. [Google Scholar]
- Dempsey, K.E.; Dorlac, W.C.; Martin, K.; Fang, R.; Fox, C.; Bennett, B.; Williams, K.; Flaherty, S. Landstuhl Regional Medical Center: Traumatic brain injury screening program. J. Trauma Nurs 2009, 16, 6–7. [Google Scholar]
- Dean, P.J.; O’Neill, D.; Sterr, A. Post-concussion syndrome: Prevalence after mild traumatic brain injury in comparison with a sample without head injury. Brain Inj 2011, 26, 14–26. [Google Scholar] [Green Version]
- Barth, J.T.; Diamond, R.; Errico, A. Mild head injury and post concussion syndrome: Does anyone really suffer? Clin. Electroencephalogr 1996, 27, 183–186. [Google Scholar]
- Beauchamp, K.; Mutlak, H.; Smith, W.R.; Shohami, E.; Stahel, P.F. Pharmacology of traumatic brain injury: Where is the “golden bullet”? Mol. Med 2008, 14, 731–740. [Google Scholar]
- Blaylock, R.L.; Maroon, J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis. Surg. Neurol. Int 2011, 2, 107. [Google Scholar]
- Roozenbeek, B.; Maas, A.I.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol 2013, 9, 231–236. [Google Scholar]
- Arand, M.; Melzner, H.; Kinzl, L.; Bruckner, U.B.; Gebhard, F. Early inflammatory mediator response following isolated traumatic brain injury and other major trauma in humans. Langenbecks Arch. Surg 2001, 386, 241–248. [Google Scholar]
- Baugh, C.M.; Stamm, J.M.; Riley, D.O.; Gavett, B.E.; Shenton, M.E.; Lin, A.; Nowinski, C.J.; Cantu, R.C.; McKee, A.C.; Stern, R.A. Chronic traumatic encephalopathy: Neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav 2012, 6, 244–254. [Google Scholar]
- Rau, T.F.; Kothiwal, A.; Zhang, L.; Ulatowski, S.; Brooks, D.M.; Cardozo-Pelaez, F.; Chopp, M.; Poulsen, D.J. Low dose methamphetamine mediates neuroprotection through a PI3K-AKT pathway. Neuropharmacology 2011, 61, 677–686. [Google Scholar]
- Bullock, R.Z.A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg 1998, 89, 507–518. [Google Scholar]
- Chan, P.H. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem. Res 2004, 29, 1943–1949. [Google Scholar]
- Faden, A.I. Neuroprotection and traumatic brain injury: Theoretical option or realistic proposition. Curr. Opin. Neurol 2002, 15, 707–712. [Google Scholar]
- Stokes, G.S.; Marwood, J.F. Review of the use of alpha-adrenoceptor antagonists in hypertension. Methods Find Exp. Clin. Pharmacol 1984, 6, 197–204. [Google Scholar]
- Strosberg, A.D. Structure, function, and regulation of adrenergic receptors. Protein. Sci 1993, 2, 1198–1209. [Google Scholar]
- Te, A.E. A modern rationale for the use of phenoxybenzamine in urinary tract disorders and other conditions. Clin. Ther 2002, 24, 851–861, discussion 837. [Google Scholar]
- Hamilton, C.A.; Dalrymple, H.W.; Reid, J.L.; Sumner, D.J. The recovery of alpha-adrenoceptor function and binding sites after phenoxybenzamine. An index of receptor turnover? Naunyn. Schmiedebergs Arch. Pharmacol 1984, 325, 34–41. [Google Scholar]
- Rau, T.F.; Kothiwal, A.S.; Rova, A.R.; Brooks, D.M.; Poulsen, D.J. Treatment with low-dose methamphetamine improves behavioral and cognitive function after severe traumatic brain injury. J. Trauma Acute. Care Surg 2012, 73, S165–S172. [Google Scholar]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process 2012, 13, 93–110. [Google Scholar]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci 2012, 14, 128–142. [Google Scholar]
- Shultz, S.R.; Bao, F.; Omana, V.; Chiu, C.; Brown, A.; Cain, D.P. Repeated mild lateral fluid percussion brain injury in the rat causes cumulative long-term behavioral impairments, neuroinflammation, and cortical loss in an animal model of repeated concussion. J. Neurotrauma 2012, 29, 281–294. [Google Scholar]
- Caine, M.; Perlberg, S.; Meretyk, S. A placebo-controlled double-blind study of the effect of phenoxybenzamine in benign prostatic obstruction. Br. J. Urol 1978, 50, 551–554. [Google Scholar]
- Buchweitz, E.; Weiss, H.R. Effects of phenoxybenzamine or N-methyl chlorpromazine on regional cerebral blood flow: Comparison of central and peripheral alpha adrenergic receptor antagonism. J. Pharmacol. Exp. Ther 1982, 223, 322–326. [Google Scholar]
- Hamill, R.W.; Woolf, P.D.; McDonald, J.V.; Lee, L.A.; Kelly, M. Catecholamines predict outcome in traumatic brain injury. Ann. Neurol 1987, 21, 438–443. [Google Scholar]
- Tran, T.Y.; Dunne, I.E.; German, J.W. Beta blockers exposure and traumatic brain injury: A literature review. Neurosurg. Focus 2008, 25, E8. [Google Scholar]
- Woolf, P.D.; Hamill, R.W.; Lee, L.A.; Cox, C.; McDonald, J.V. The predictive value of catecholamines in assessing outcome in traumatic brain injury. J. Neurosurg 1987, 66, 875–882. [Google Scholar]
- Woolf, P.D.; Hamill, R.W.; Lee, L.A.; McDonald, J.V. Free and total catecholamines in critical illness. Am. J. Physiol 1988, 254, E287–E291. [Google Scholar]
- Kobori, N.; Hu, B.; Dash, P.K. Altered adrenergic receptor signaling following traumatic brain injury contributes to working memory dysfunction. Neuroscience 2011, 172, 293–302. [Google Scholar]
- Cimino, M.; Weiss, B. Characteristics of the binding of phenoxybenzamine to calmodulin. Biochem. Pharmacol 1988, 37, 2739–2745. [Google Scholar]
- Yang, K.; Taft, W.C.; Dixon, C.E.; Todaro, C.A.; Yu, R.K.; Hayes, R.L. Alterations of protein kinase C in rat hippocampus following traumatic brain injury. J. Neurotrauma 1993, 10, 287–295. [Google Scholar]
- Tomycz, N.D.; Friedlander, R.M. α-1 Adrenergic receptor signaling linked to memory dysfunction following brain trauma. Neurosurgery 2011, 68, N16–N18. [Google Scholar]
- Vest, R.S.; O’Leary, H.; Coultrap, S.J.; Kindy, M.S.; Bayer, K.U. Effective post-insult neuroprotection by a novel Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) inhibitor. J. Biol. Chem 2010, 285, 20675–20682. [Google Scholar]
- Zhang, M.; Shan, H.; Gu, Z.; Wang, D.; Wang, T.; Wang, Z.; Tao, L. Increased expression of calcium/calmodulin-dependent protein kinase type II subunit delta after rat traumatic brain injury. J. Mol. Neurosci 2012, 46, 631–643. [Google Scholar]
- Semple, B.D.; Frugier, T.; Morganti-Kossmann, M.C. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): Evidence from severe TBI patients and CCL2−/− mice. J. Cereb. Blood Flow Metab 2010, 30, 769–782. [Google Scholar]
- Semple, B.D.; Frugier, T.; Morganti-Kossmann, M.C. CCL2 modulates cytokine production in cultured mouse astrocytes. J. Neuroinflammation 2010, 7, 67. [Google Scholar]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of chemokines in CNS health and pathology: A focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab 2010, 30, 459–473. [Google Scholar]
- Sozzani, S.; Rieppi, M.; Locati, M.; Zhou, D.; Bussolino, F.; Proost, P.; van Damme, J.; Mantovani, A. Synergism between platelet activating factor and C–C chemokines for arachidonate release in human monocytes. Biochem. Biophys. Res. Commun 1994, 199, 761–766. [Google Scholar]
- Sozzani, S.; Zhou, D.; Locati, M.; Rieppi, M.; Proost, P.; Magazin, M.; Vita, N.; van Damme, J.; Mantovani, A. Receptors and transduction pathways for monocyte chemotactic protein-2 and monocyte chemotactic protein-3. Similarities and differences with MCP-1. J. Immunol 1994, 152, 3615–3622. [Google Scholar]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar]
- Rhodes, J.K.; Sharkey, J.; Andrews, P.J. The temporal expression, cellular localization, and inhibition of the chemokines MIP-2 and MCP-1 after traumatic brain injury in the rat. J. Neurotrauma 2009, 26, 507–525. [Google Scholar]
- Prodjosudjadi, W.; Gerritsma, J.S.; Klar-Mohamad, N.; Gerritsen, A.F.; Brujin, J.A.; Daha, M.R.; van Es, L.A. Production and cytokine-mediated regulation of monocyte chemoattractant protein-1 by human proximal tubular epithelial cells. Kidney. Int 1995, 48, 1477–1486. [Google Scholar]
- Prodjosudjadi, W.; Gerritsma, J.S.; van Es, L.A.; Daha, M.R.; Bruijn, J.A. Monocyte chemoattractant protein-1 in normal and diseased human kidneys: An immunohistochemical analysis. Clin. Nephrol 1995, 44, 148–155. [Google Scholar]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci 2007, 8, 57–69. [Google Scholar]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar]
- Lloyd, E.; Somera-Molina, K.; van Eldik, L.J.; Watterson, D.M.; Wainwright, M.S. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J. Neuroinflammation 2008, 5, 28. [Google Scholar]
- Brown, G.C.; Neher, J.J. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol. Neurobiol 2010, 41, 242–247. [Google Scholar]
- Li, B.; Mahmood, A.; Lu, D.; Wu, H.; Xiong, Y.; Qu, C.; Chopp, M. Simvastatin attenuates microglial cells and astrocyte activation and decreases interleukin-1beta level after traumatic brain injury. Neurosurgery 2009, 65, 179–185, Discussion 185–186. [Google Scholar]
- Li, G.Z.; Zhang, Y.; Zhao, J.B.; Wu, G.J.; Su, X.F.; Hang, C.H. Expression of myeloid differentiation primary response protein 88 (Myd88) in the cerebral cortex after experimental traumatic brain injury in rats. Brain Res 2011, 1396, 96–104. [Google Scholar]
- Ling, H.P.; Li, W.; Zhou, M.L.; Tang, Y.; Chen, Z.R.; Hang, C.H. Expression of intestinal myeloid differentiation primary response protein 88 (Myd88) following experimental traumatic brain injury in a mouse model. J. Surg. Res 2013, 179, e227–e234. [Google Scholar]
- Janssens, S.; Beyaert, R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem. Sci 2002, 27, 474–482. [Google Scholar]
- Selkirk, J.V.; Stiefel, T.H.; Stone, I.M.; Naeve, G.S.; Foster, A.C.; Poulsen, D.J. Over-expression of the human EAAT2 glutamate transporter within neurons of mouse organotypic hippocampal slice cultures leads to increased vulnerability of CA1 pyramidal cells. Eur. J. Neurosci 2005, 21, 2291–2205. [Google Scholar]
- Noraberg, J.; Kristensen, B.W.; Zimmer, J. Markers for neuronal degeneration in organotypic slice cultures. Brain Res. Brain Res. Protoc 1999, 3, 278–290. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Saline | Phenoxybenzamine | ||
|---|---|---|---|---|
| Fold Change | p-value | Fold Change | p-value | |
| CCL2 | 11.12 | 0.004 | 3.34 | 0.92 |
| CXCL12 | 1.58 | 0.04 | 1.02 | 0.55 |
| Fos | 1.46 | 0.23 | −1.64 | 0.08 |
| IL1β | 4.58 | 0.005 | 1.53 | 0.71 |
| Gadd45g | 2.22 | 0.69 | 1.02 | 0.16 |
| Myd88 | 3.03 | 0.0001 | 1.27 | 0.20 |
| Pdgfα | 1.21 | 0.10 | −1.28 | 0.09 |
| NTS | 1.12 | 0.60 | 1.23 | 0.19 |
| Rbp2 | 3.96 | 0.00008 | 1.58 | 0.03 |
| CRH | −1.66 | 0.002 | −1.56 | 0.003 |
| GRIA4 | 1.47 | 0.005 | −1.14 | 0.50 |
| GRIA2 | 1.11 | 0.42 | −1.17 | 0.19 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rau, T.F.; Kothiwal, A.; Rova, A.; Rhoderick, J.F.; Poulsen, D.J. Phenoxybenzamine Is Neuroprotective in a Rat Model of Severe Traumatic Brain Injury. Int. J. Mol. Sci. 2014, 15, 1402-1417. https://doi.org/10.3390/ijms15011402
Rau TF, Kothiwal A, Rova A, Rhoderick JF, Poulsen DJ. Phenoxybenzamine Is Neuroprotective in a Rat Model of Severe Traumatic Brain Injury. International Journal of Molecular Sciences. 2014; 15(1):1402-1417. https://doi.org/10.3390/ijms15011402
Chicago/Turabian StyleRau, Thomas F., Aakriti Kothiwal, Annela Rova, Joseph F. Rhoderick, and David J. Poulsen. 2014. "Phenoxybenzamine Is Neuroprotective in a Rat Model of Severe Traumatic Brain Injury" International Journal of Molecular Sciences 15, no. 1: 1402-1417. https://doi.org/10.3390/ijms15011402