EGF Potentiation of VEGF Production Is Cell Density Dependent in H292 EGFR Wild Type NSCLC Cell Line

Abstract
:1. Introduction
2. Results and Discussion
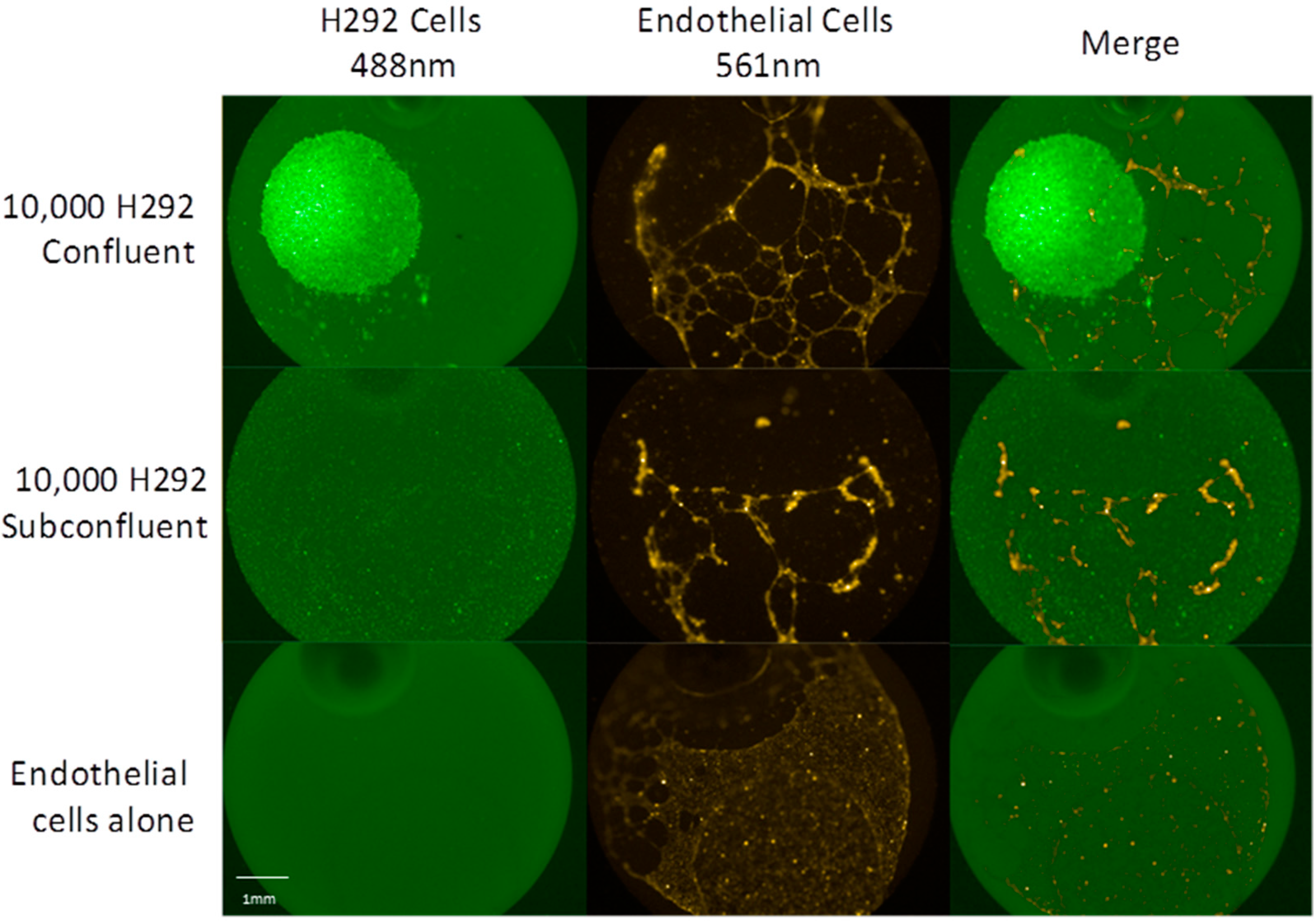
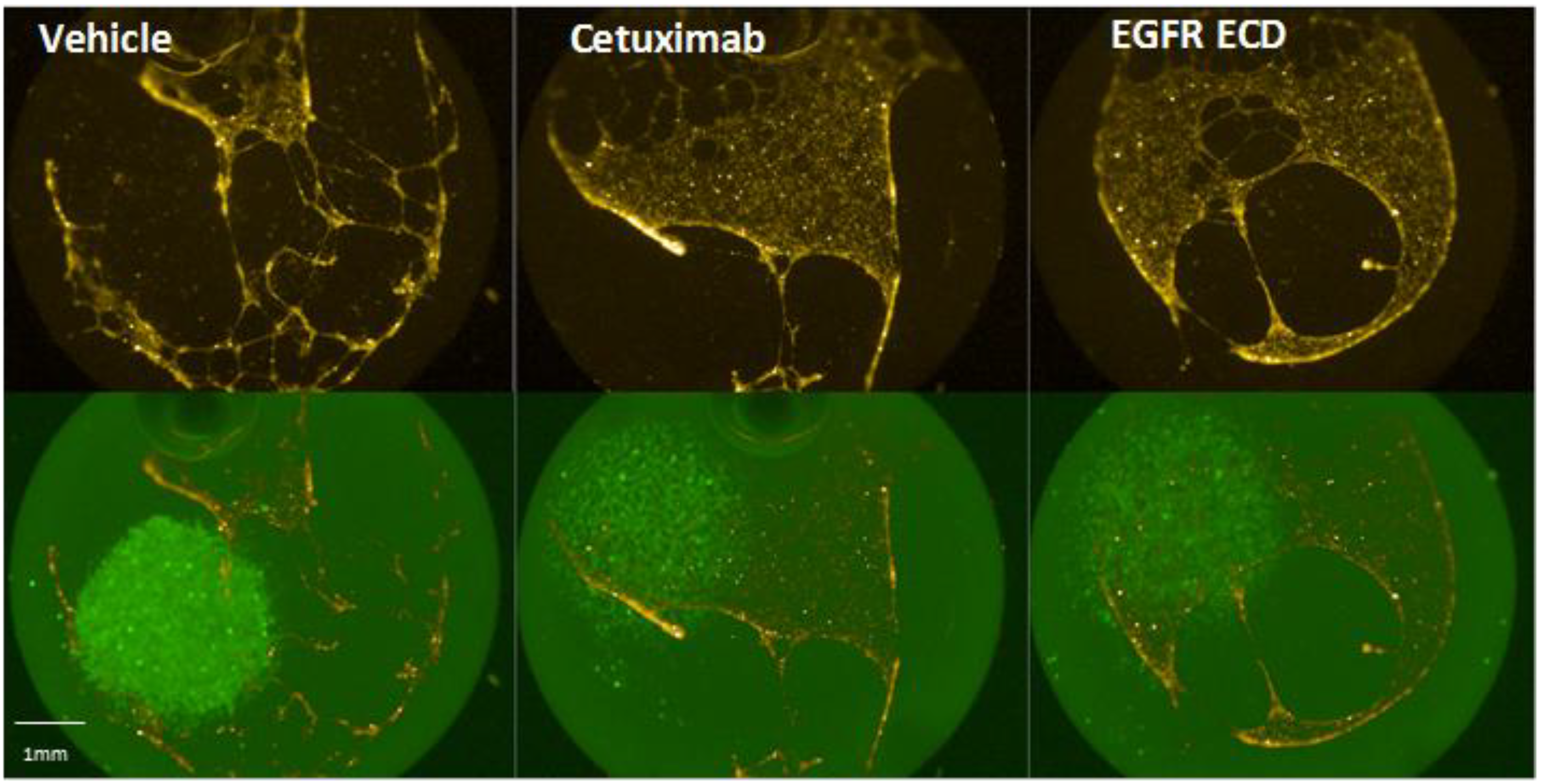
2.1. Dense Cell Spots Promote Angiogenesis to a Greater Degree than Sparse Cell Lawns

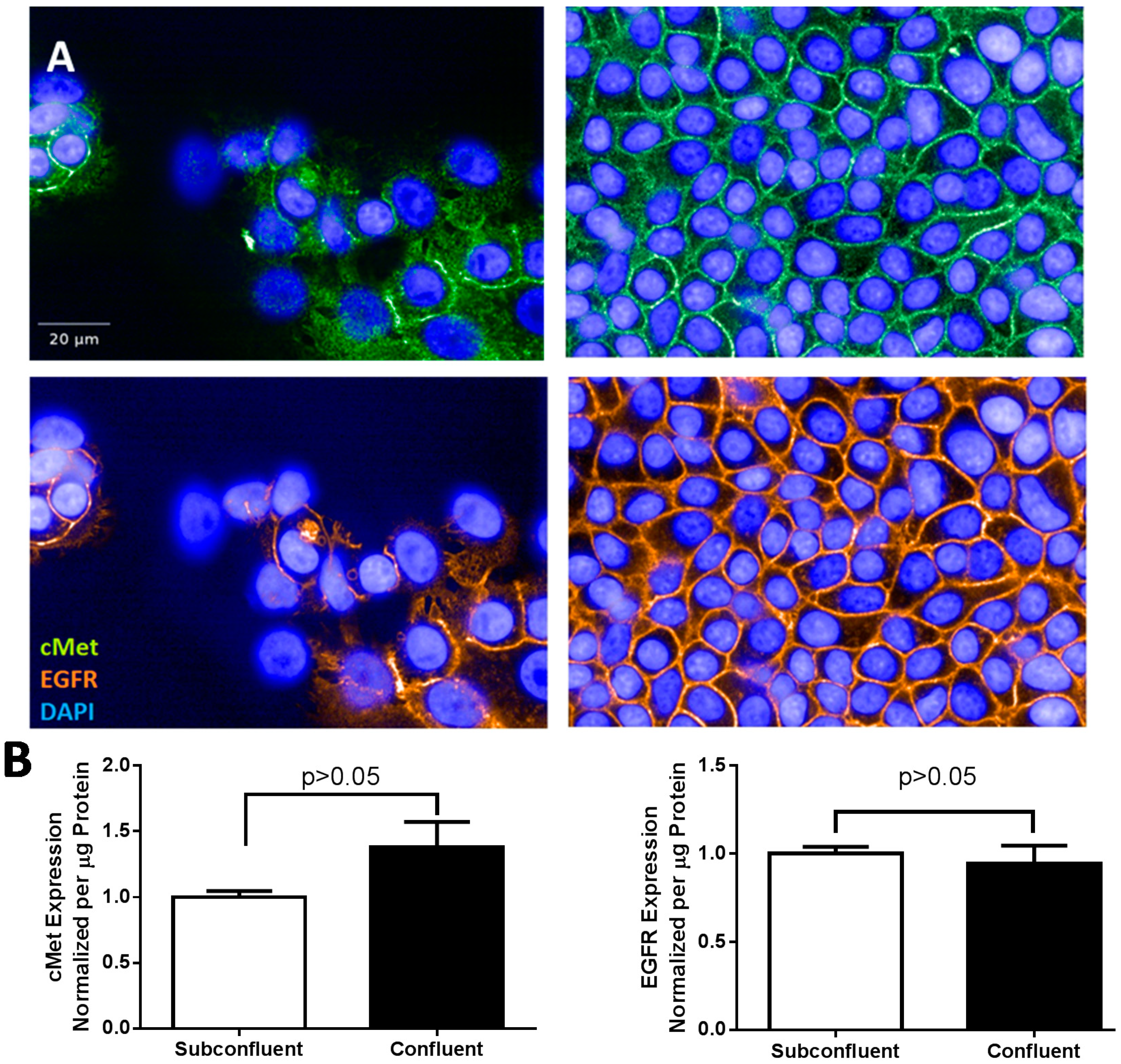
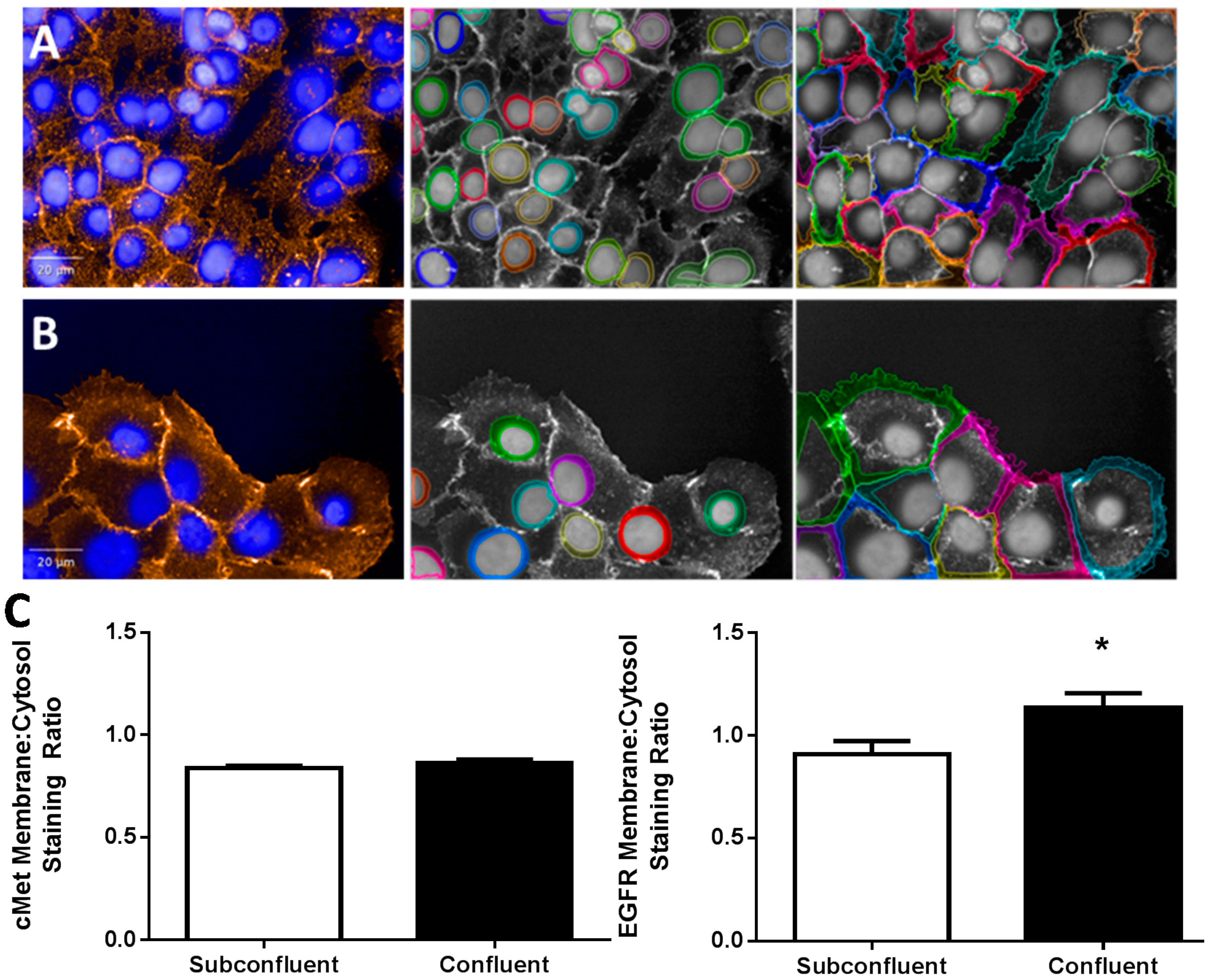
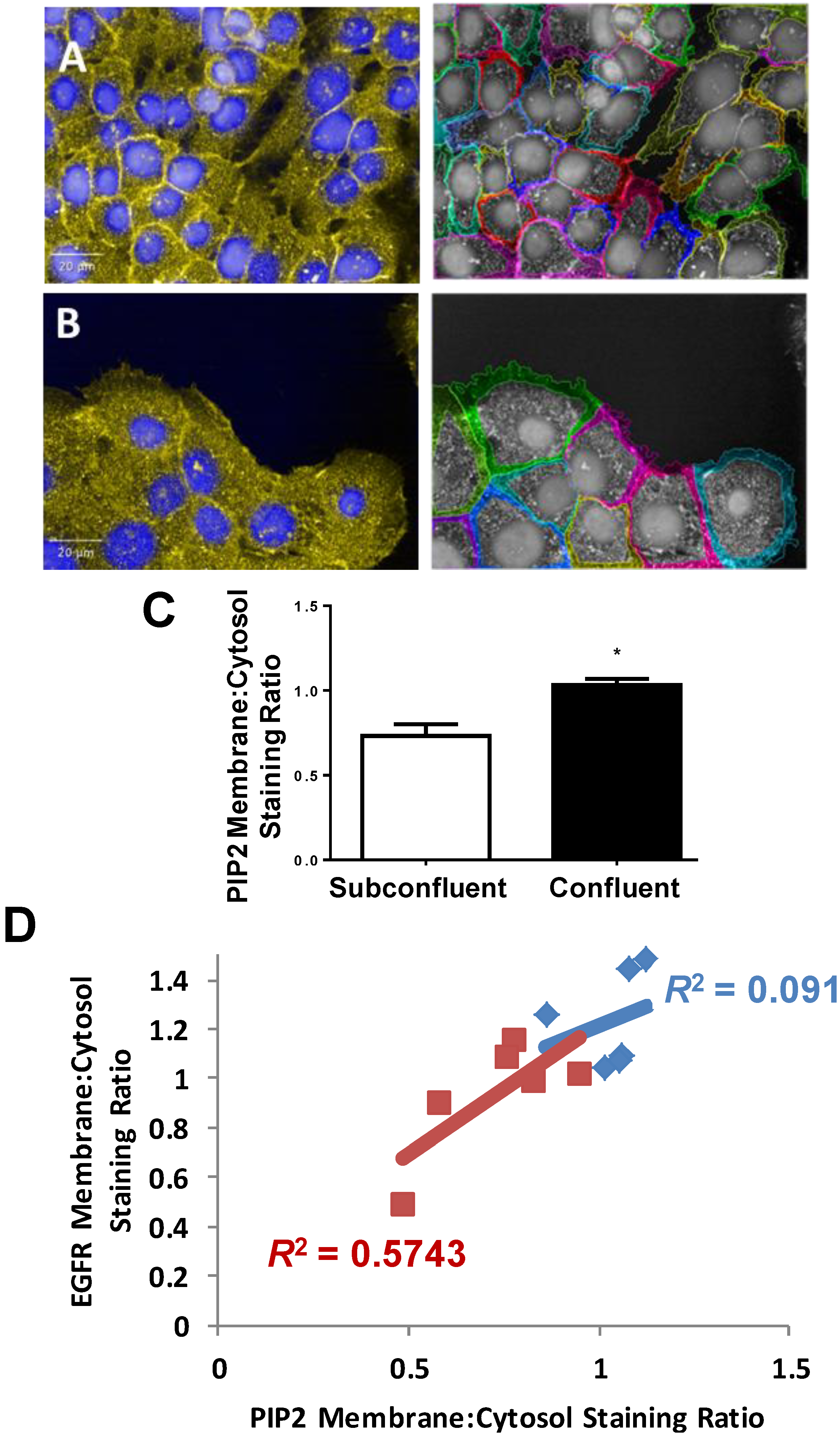
2.2. EGFR Plasma Membrane Localization Is Enhanced in Confluent H292 Cells

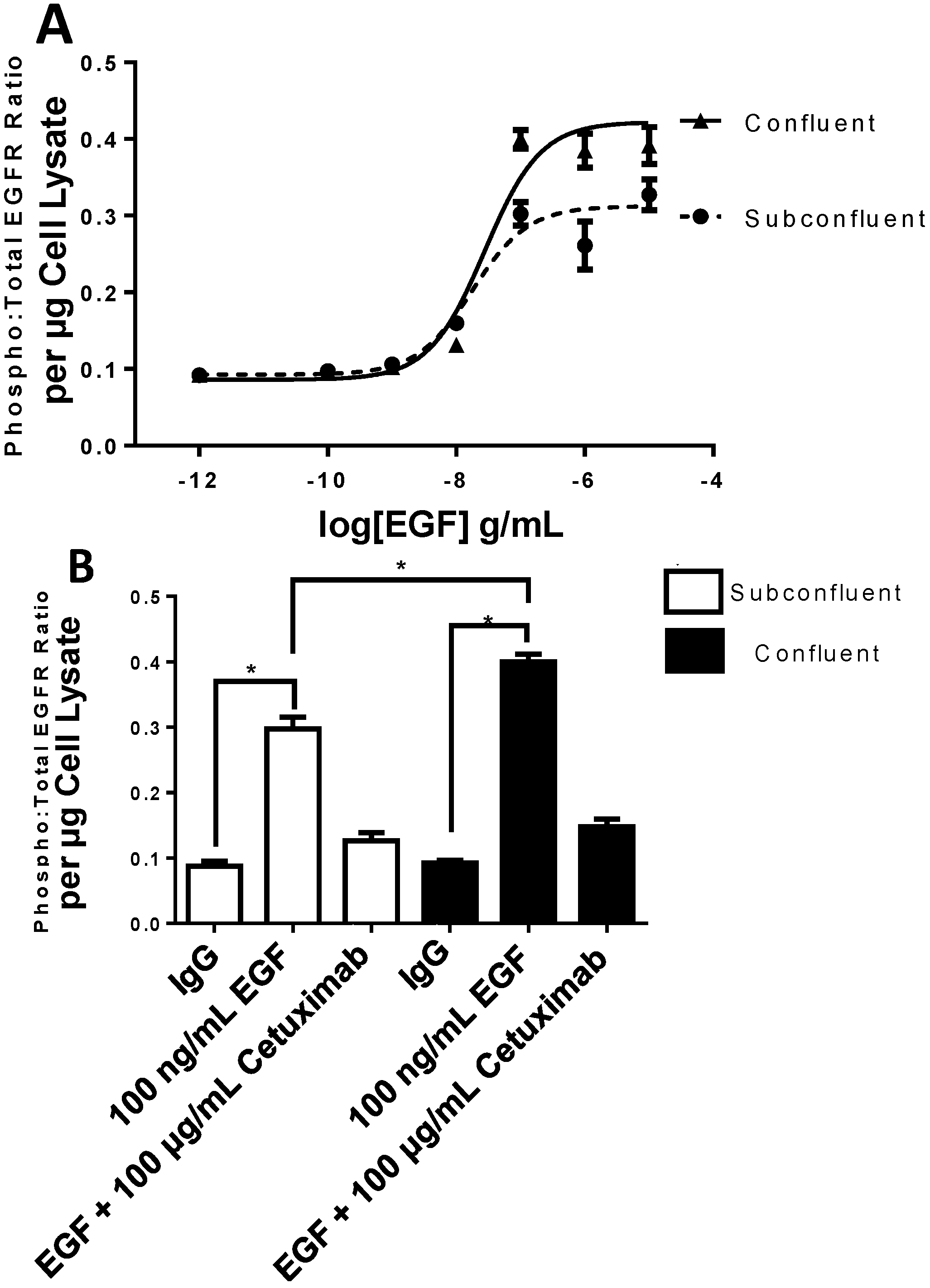
2.3. EGFR Phosphorylation Is Enhanced in Confluent Cells


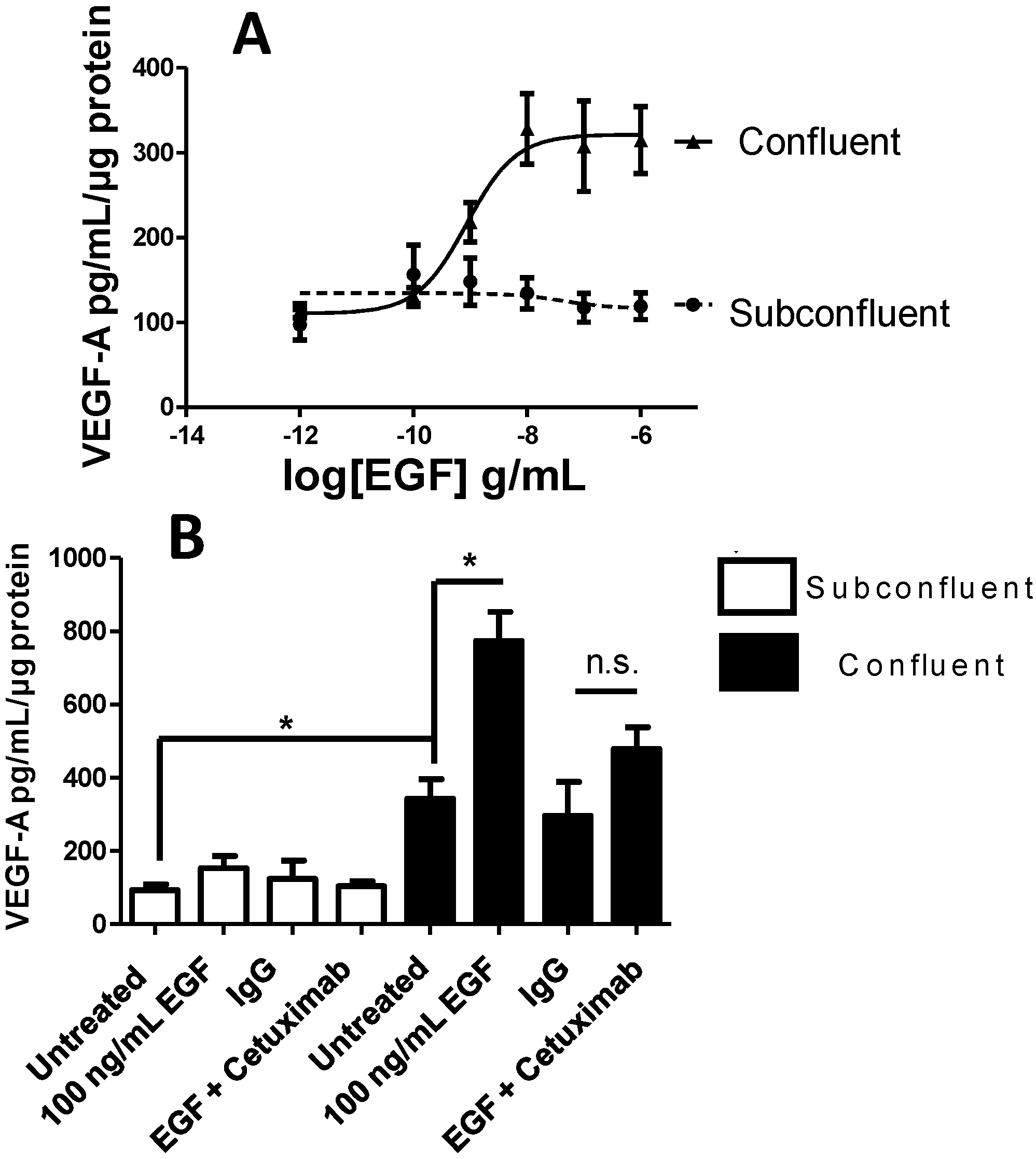
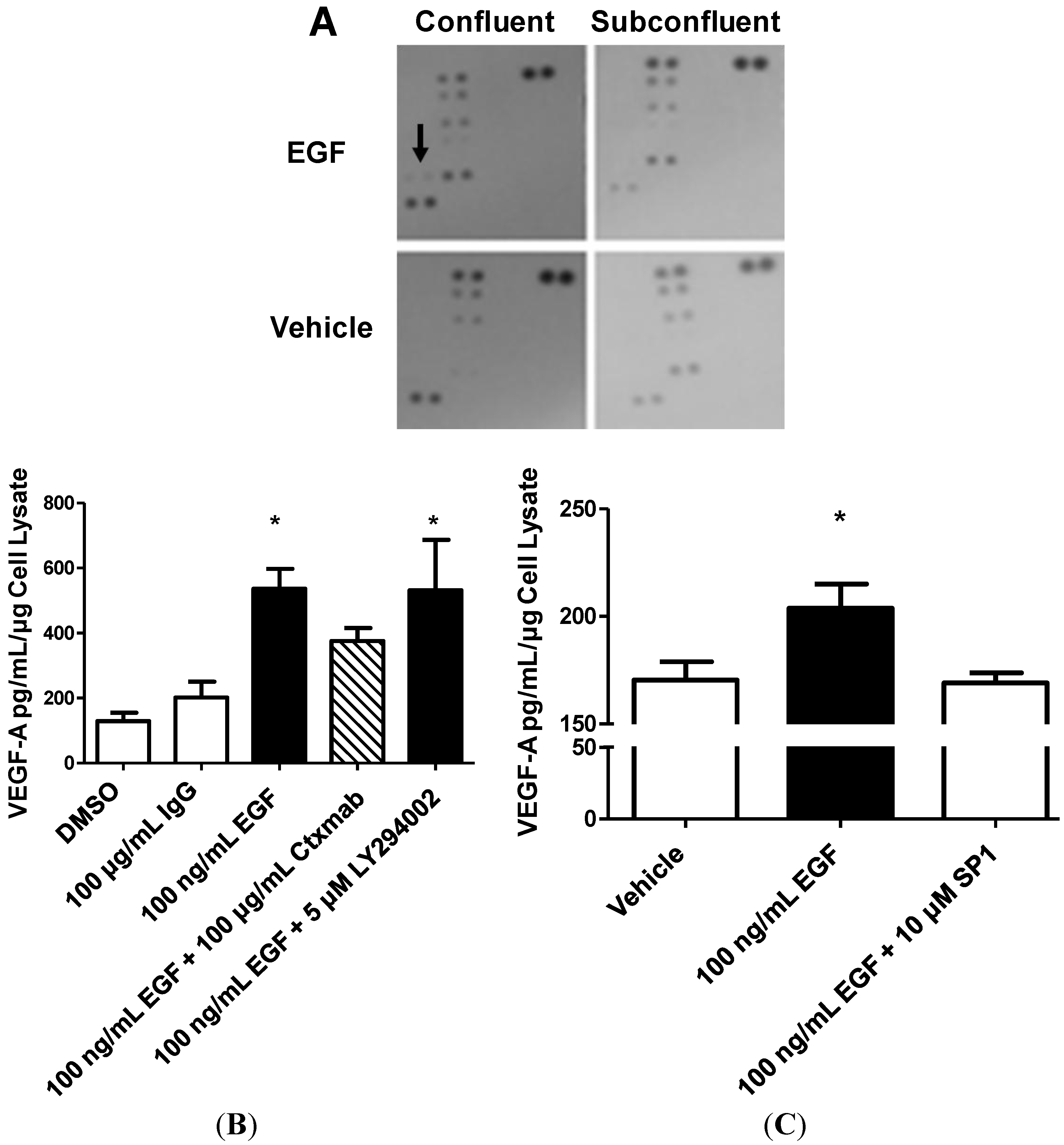
2.4. Vascular Endothelial Growth Factor (VEGF) A Transcription and Secretion Is Stimulated by Epidermal Growth Factor (EGF) Treatment in Confluent Cells

2.5. Angiogenesis Promoted by Dense H292 Cell Spots Is EGFR Dependent

2.6. STAT3, but not PI3K, Mediates EGFR-Stimulated VEGF-A Secretion



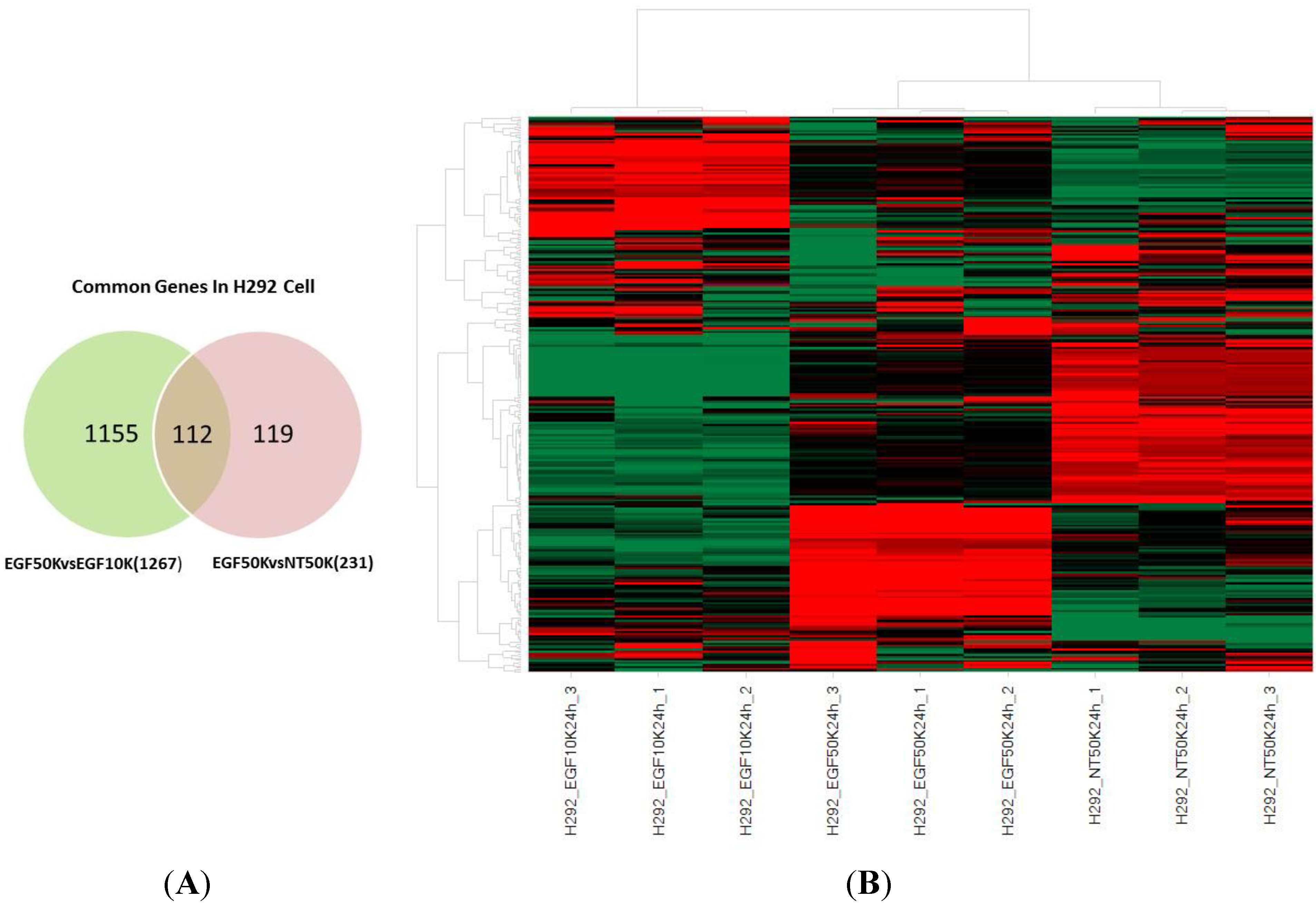
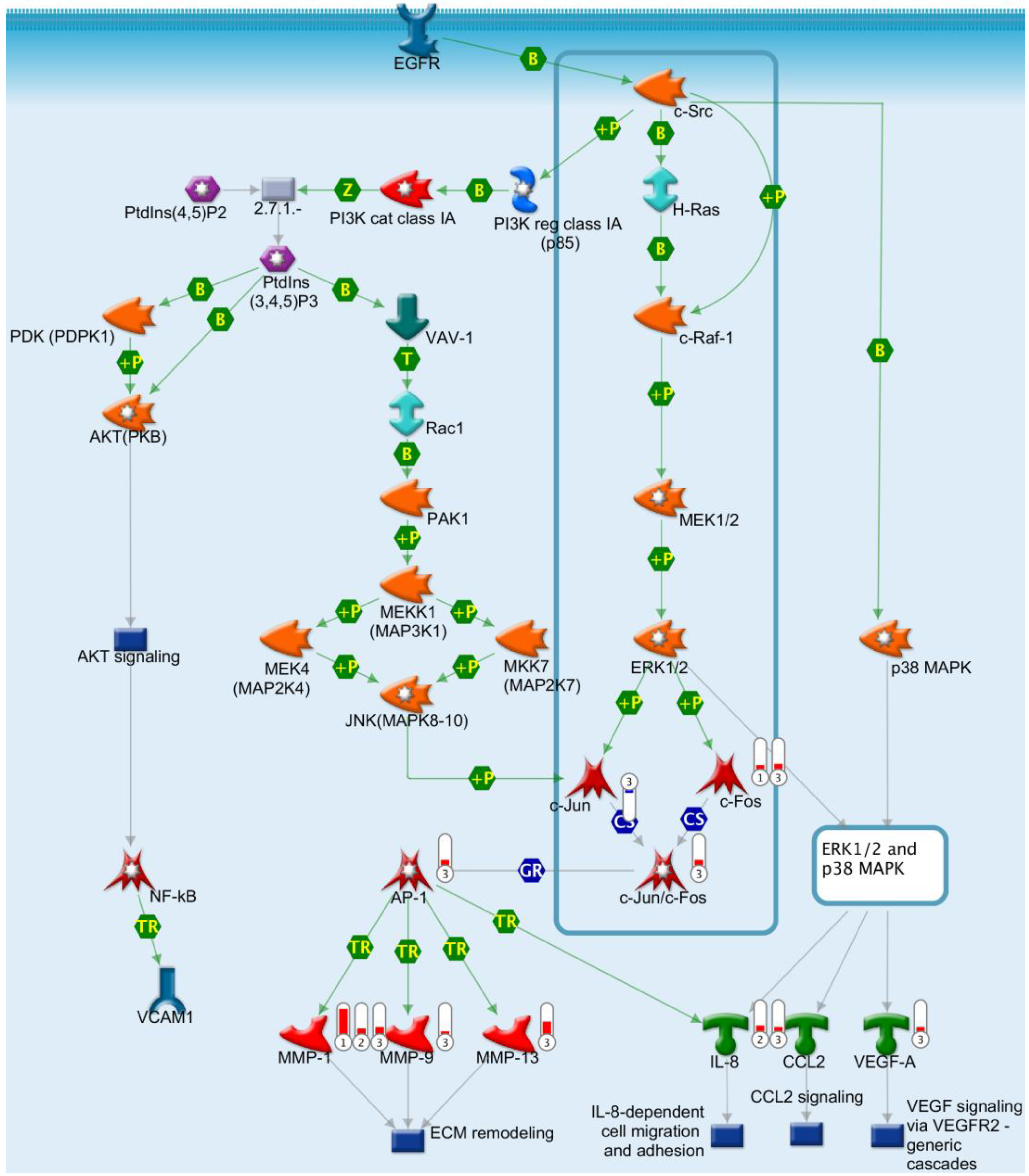
2.7. Gene Chip Analysis and Pathway Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Control Group (A) | Comparison Group (B) | Number of Differentially Regulated Genes (A vs. B) |
|---|---|---|---|
| 1 | Confluent + saline (NT50K 24 h) | Confluent + EGF (EGF50K 24 h) | 231 |
| 2 | Subconfluent + saline (NT10K 24 h) | Subconfluent + EGF (EGF10K 24 h) | 86 |
| 3 | Subconfluent + EGF (EGF10K 24 h) | Confluent + EGF (EGF50K 24 h) | 1267 |

2.8. Potentiation of EGFR-Induced VEGF mRNA Production through Increased Cell Density Is Exclusive to H292 Cells
3. Experimental Section
3.1. Reagents & Cells
3.2. Angiogenesis Assay
3.3. Protein and Lipid Staining
3.4. EGFR Phosphorylation
3.5. VEGF Quantification
3.6. STAT3 Phosphorylation
3.7. Gene Expression
3.8. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reck, M.; Heigener, D.F.; Mok, T.; Soria, J.-C.; Rabe, K.F. Management of non-small-cell lung cancer: Recent developments. Lancet 2013, 382, 709–719. [Google Scholar]
- Pirker, R.; Herth, F.J.; Kerr, K.M.; Filipits, M.; Taron, M.; Gandara, D.; Hirsch, F.R.; Grunenwald, D.; Popper, H.; Smit, E.; et al. Consensus for EGFR mutation testing in non-small cell lung cancer: Results from a European workshop. J. Thorac. Oncol. 2010, 5, 1706–1713. [Google Scholar]
- Zucali, P.A.; Ruiz, M.G.; Giovannetti, E.; Destro, A.; Varella-Garcia, M.; Floor, K.; Ceresoli, G.L.; Rodriguez, J.A.; Garassino, I.; Comoglio, P.; et al. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Ann. Oncol. 2008, 19, 1605–1612. [Google Scholar] [CrossRef]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Loriot, Y.; Mordant, P.; Deutsch, E.; Olaussen, K.A.; Soria, J.C. Are RAS mutations predictive markers of resistance to standard chemotherapy? Nat. Rev. Clin. Oncol. 2009, 6, 528–534. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Boch, C.; Kollmeier, J.; Roth, A.; Stephan-Falkenau, S.; Misch, D.; Gruning, W.; Bauer, T.T.; Mairinger, T. The frequency of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC): Routine screening data for central Europe from a cohort study. BMJ Open 2013. [Google Scholar] [CrossRef]
- Gullick, W.J.; Downward, J.; Parker, P.J.; Whittle, N.; Kris, R.; Schlessinger, J.; Ullrich, A.; Waterfield, M.D. The structure and function of the epidermal growth factor receptor studied by using antisynthetic peptide antibodies. Proc. R. Soc. Lond. B Biol. Sci. 1985, 226, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; McGraw, T.E. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 2004, 5, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Nyati, M.K.; Morgan, M.A.; Feng, F.Y.; Lawrence, T.S. Integration of EGFR inhibitors with radiochemotherapy. Nat. Rev. Cancer 2006, 6, 876–885. [Google Scholar] [CrossRef]
- Tsai, J.-C.; Goldman, C.K.; Gillespie, G.Y. Vascular endothelial growth factor in human glioma cell lines: Induced secretion by EGF, PDGF-BB, and bFGF. J. Neurosurg. 1995, 82, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Keller, G.A.; Ferrara, N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell 1993, 4, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef]
- Robinson, C.J.; Stringer, S.E. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 2001, 114, 853–865. [Google Scholar] [PubMed]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Brekken, R.A.; Overholser, J.P.; Stastny, V.A.; Waltenberger, J.; Minna, J.D.; Thorpe, P.E. Selective inhibition of vascular endothelial growth factor (VEGF) receptor 2 (KDR/Flk-1) activity by a monoclonal anti-VEGF antibody blocks tumor growth in mice. Cancer Res. 2000, 60, 5117–5124. [Google Scholar]
- Swat, A.; Dolado, I.; Rojas, J.M.; Nebreda, A.R. Cell density-dependent inhibition of epidermal growth factor receptor signaling by p38α mitogen-activated protein kinase via Sprouty2 downregulation. Mol. Cell. Biol. 2009, 29, 3332–3343. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Kushiro, K.; Graham, N.A.; Asthagiri, A.R. Tunable interplay between epidermal growth factor and cell–cell contact governs the spatial dynamics of epithelial growth. Proc. Natl. Acad. Sci. USA 2009, 106, 11149–11153. [Google Scholar] [CrossRef] [PubMed]
- Sawano, A.; Takayama, S.; Matsuda, M.; Miyawaki, A. Lateral propagation of EGF signaling after local stimulation is dependent on receptor density. Dev. Cell 2002, 3, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.B.; Pedersen, M.W.; Stockhausen, M.T.; Grandal, M.V.; van Deurs, B.; Poulsen, H.S. Activation of the EGFR gene target EphA2 inhibits epidermal growth factor-induced cancer cell motility. Mol. Cancer Res. MCR 2007, 5, 283–293. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranayhossaini, D.J.; Lu, J.; Mabus, J.; Gervais, A.; Lingham, R.B.; Fursov, N. EGF Potentiation of VEGF Production Is Cell Density Dependent in H292 EGFR Wild Type NSCLC Cell Line. Int. J. Mol. Sci. 2014, 15, 17686-17704. https://doi.org/10.3390/ijms151017686
Ranayhossaini DJ, Lu J, Mabus J, Gervais A, Lingham RB, Fursov N. EGF Potentiation of VEGF Production Is Cell Density Dependent in H292 EGFR Wild Type NSCLC Cell Line. International Journal of Molecular Sciences. 2014; 15(10):17686-17704. https://doi.org/10.3390/ijms151017686
Chicago/Turabian StyleRanayhossaini, Daniel J., Jin Lu, John Mabus, Alexis Gervais, Russell B. Lingham, and Natalie Fursov. 2014. "EGF Potentiation of VEGF Production Is Cell Density Dependent in H292 EGFR Wild Type NSCLC Cell Line" International Journal of Molecular Sciences 15, no. 10: 17686-17704. https://doi.org/10.3390/ijms151017686
APA StyleRanayhossaini, D. J., Lu, J., Mabus, J., Gervais, A., Lingham, R. B., & Fursov, N. (2014). EGF Potentiation of VEGF Production Is Cell Density Dependent in H292 EGFR Wild Type NSCLC Cell Line. International Journal of Molecular Sciences, 15(10), 17686-17704. https://doi.org/10.3390/ijms151017686