Microenvironment of Tumor-Draining Lymph Nodes: Opportunities for Liposome-Based Targeted Therapy

Abstract
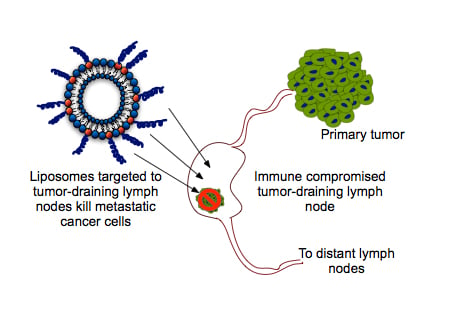
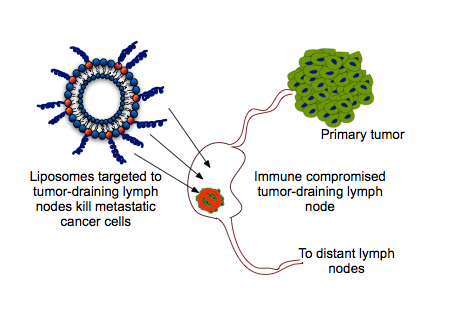
:
{kind=link}
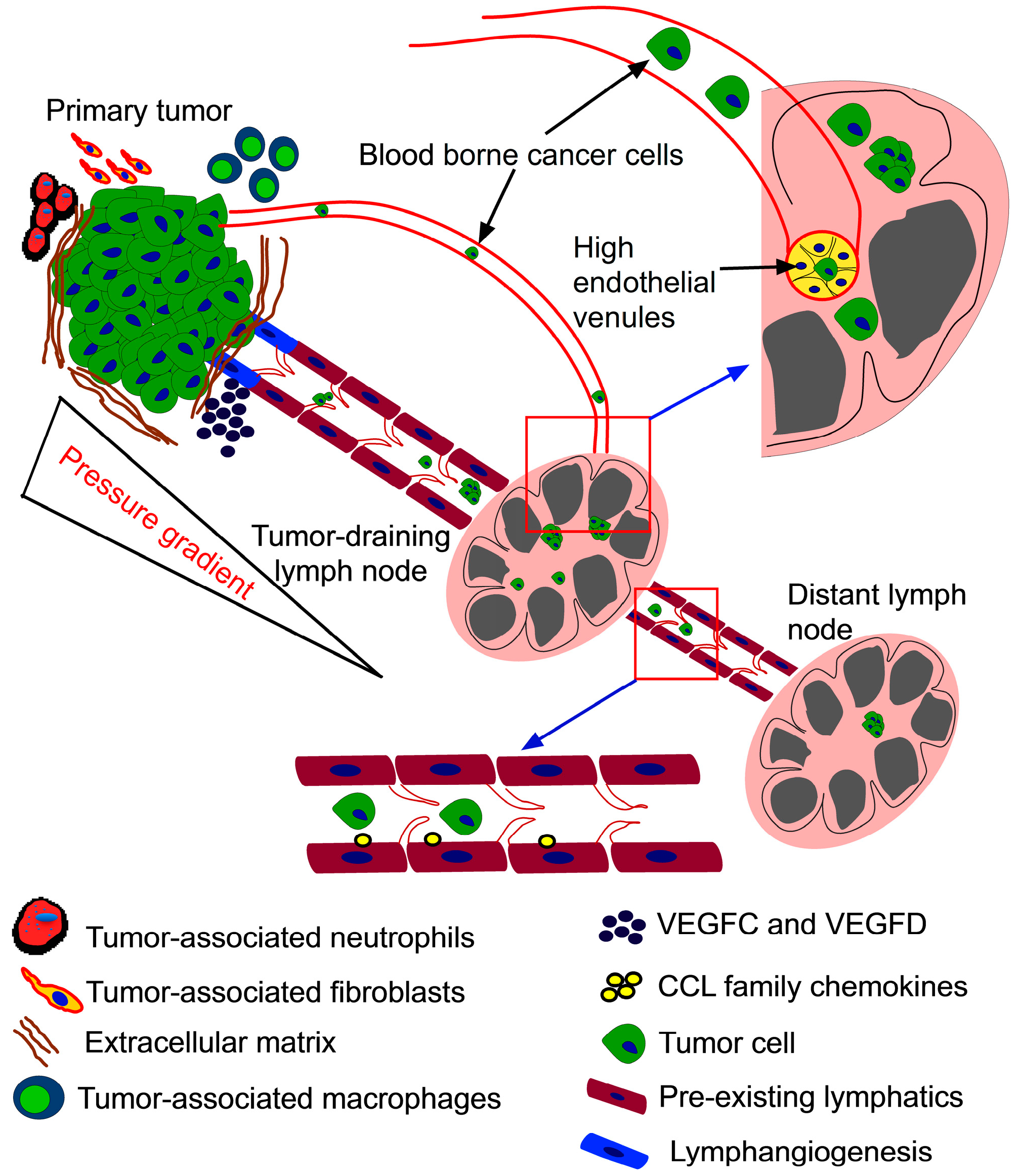{kind=link}
{kind=link}
{kind=link}
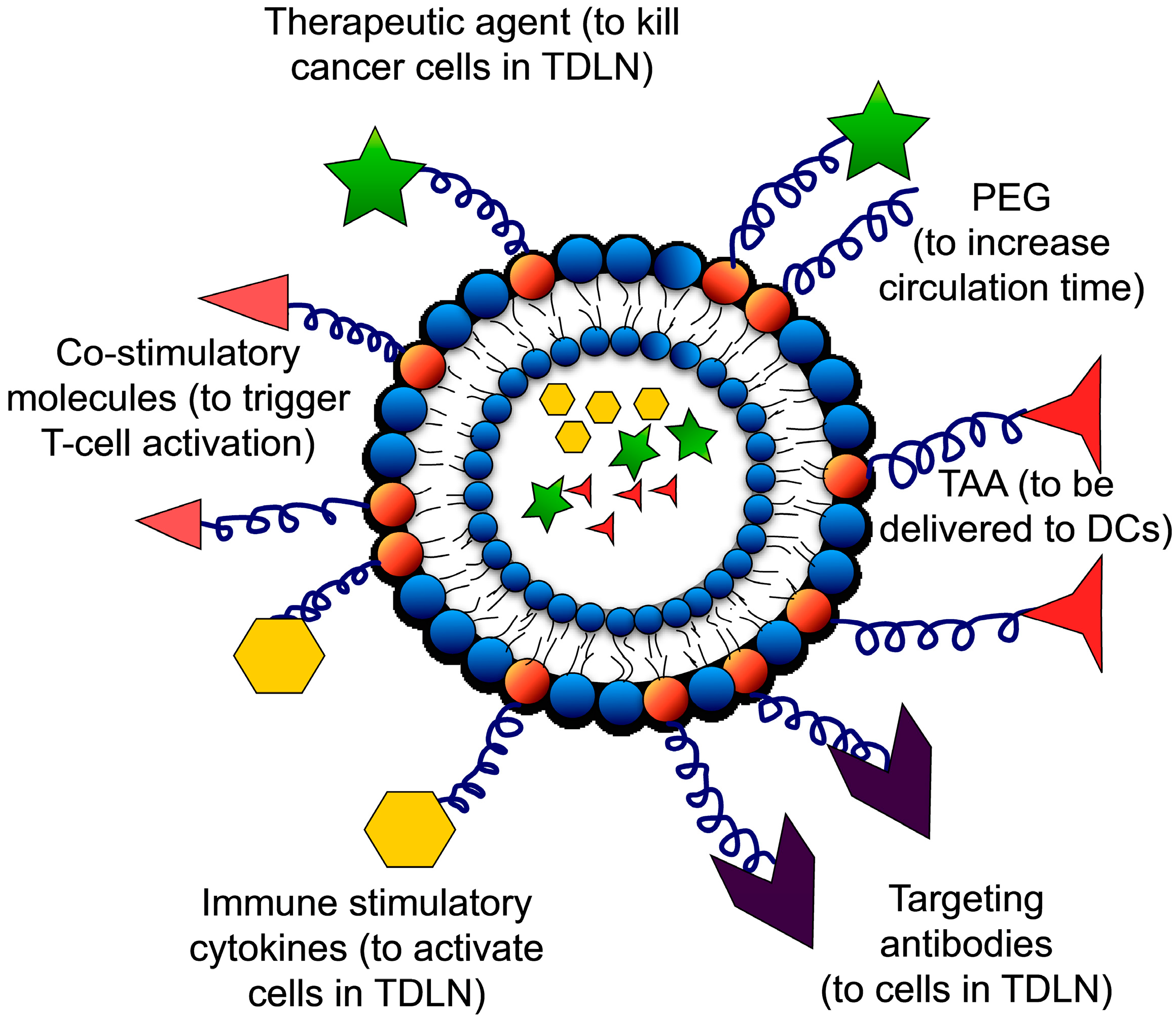{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Microenvironment of the Tumor-Draining Lymph Node


2.1. Natural Killer Cells
2.2. T-Cells
2.3. Antigen Presenting Cells
2.4. B-Cells
2.5. From Local Tolerance to Systemic Tolerance
3. Liposome-Based TDLN Targeted Immunotherapy

3.1. Factors Affecting the Delivery of Liposomes to Lymph Nodes
3.2. Delivering Liposomes to the TDLN by Targeting DC
3.3. Delivering Liposomes to the TDLN by Targeting T-Cells
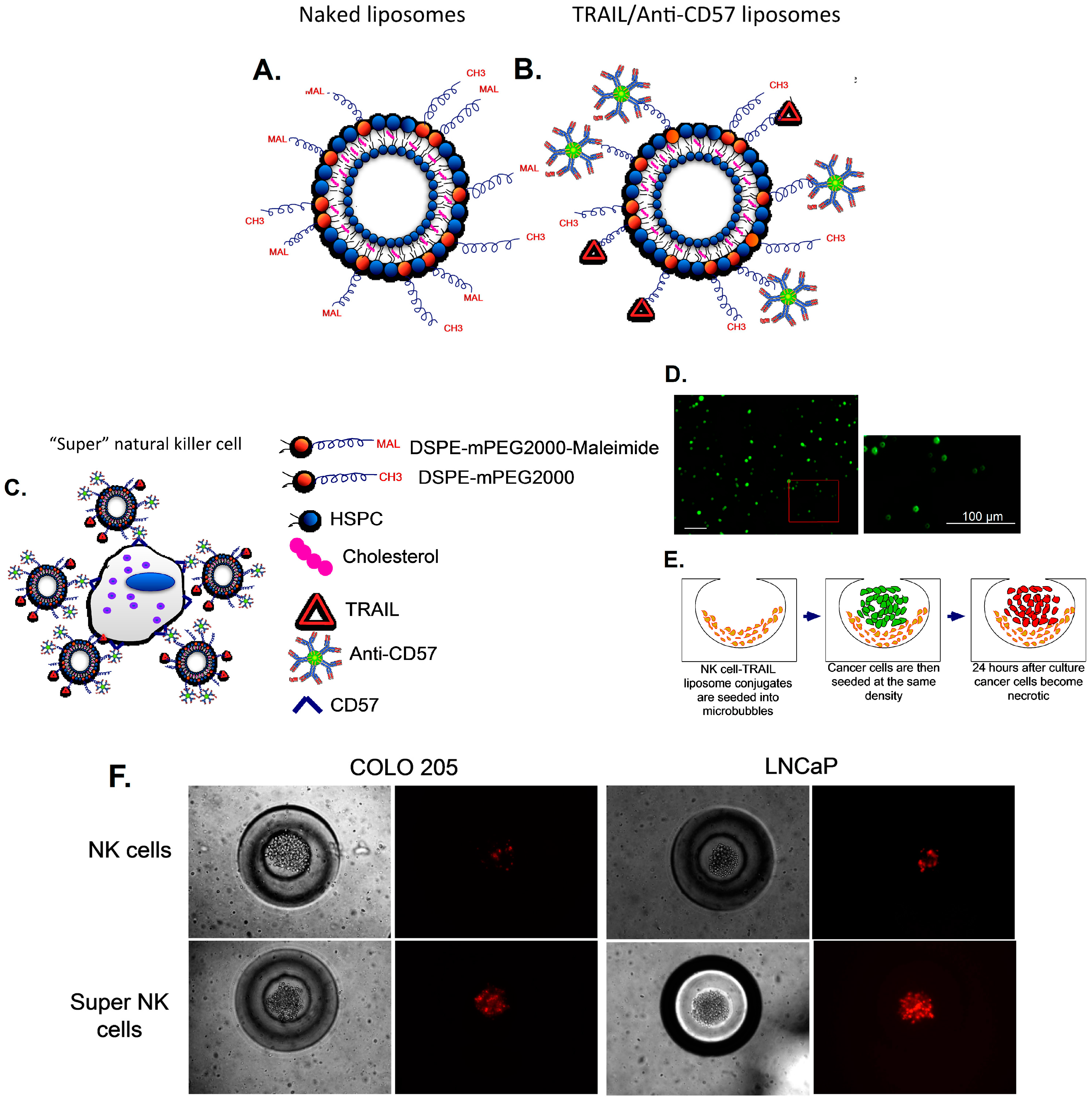
3.4. Delivering Therapeutic Liposomes to TDLN by Targeting NK Cells
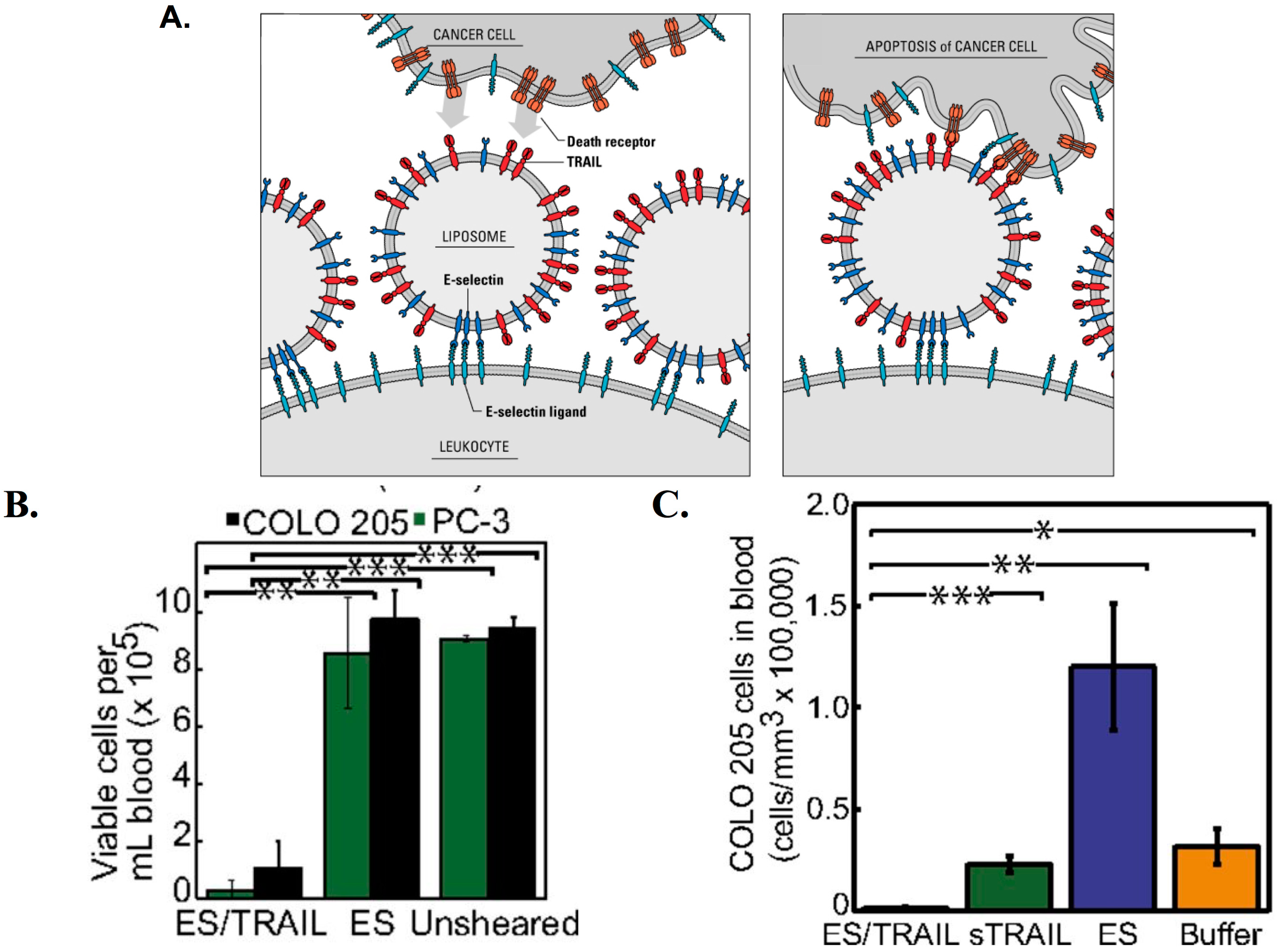
3.5. Therapeutic TRAIL-Liposomes Directed to Circulating Leukocytes


4. Concluding Remarks
Conflicts of Interest
References
- Thompson, J.F.; McCarthy, W.H.; Bosch, C.M.; O’Brien, C.J.; Quinn, M.J.; Paramaesvaran, S.; Crotty, K.; McCarthy, S.W.; Uren, R.F.; Howman-Giles, R. Sentinel lymph node status as an indicator of the presence of metastatic melanoma in regional lymph nodes. Melanoma Res. 1995, 5, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Van Trappen, P.O.; Pepper, M.S. Lymphatic dissemination of tumour cells and the formation of micrometastases. Lancet Oncol. 2002, 3, 44–52. [Google Scholar]
- Ariel, I.M. Incidence of metastases to lymph nodes from soft-tissue sarcomas. Semin. Surg. Oncol. 1988, 4, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, I.; Hilsenbeck, S.G.; Clark, G.M.; Osborne, C.K. Significance of axillary lymph node metastasis in primary breast cancer. J. Clin. Oncol. 1999, 17, 2334–2340. [Google Scholar] [PubMed]
- Ollila, D.B.; Stitzenberg, K.B. Breast cancer sentinel node metastases: Histopathologic detection and clinical significance. Cancer Control 2001, 8, 407–414. [Google Scholar] [PubMed]
- Massucco, P.; Ribero, D.; Sgotto, E.; Mellano, A.; Muratore, A.; Capussotti, L. Prognostic significance of lymph node metastases in pancreatic head cancer treated with extended lymphadenectomy: Not just a matter of numbers. Ann. Surg. Oncol. 2009, 16, 3323–3332. [Google Scholar] [CrossRef]
- Gervasi, L.A.; Mata, J.; Easley, J.D.; Wilbanks, J.H.; Seale-Hawkins, C.; Carlton, C.E.; Scardino, P.T. Prognostic significance of lymph nodal metastases in prostate cancer. J. Urol. 1989, 142, 332–336. [Google Scholar] [PubMed]
- Saito, H.; Fukumoto, Y.; Osaki, T.; Fukuda, K.; Tatebe, S.; Tsujitani, S.; Ikeguchi, M. Prognostic significance of level and number of lymph node metastases in patients with gastric cancer. Ann. Surg. Oncol. 2007, 14, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Willard-Mack, C.L. Normal structure, function, and histology of lymph nodes. Toxicol. Pathol. 2006, 34, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Von Andrian, U.H.; Mempel, T.R. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 2003, 3, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.P.L.; Nakakura, E.K.; Pollock, R.; Choti, M.A.; Morton, D.L.; Henner, W.D.; Lal, A.; Pillai, R.; Clark, O.H.; Cady, B. Unique patterns of metastases in common and rare types of malignancy. J. Surg. Oncol. 2011, 103, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Polyak, K. Microenvironmental regulation of cancer development. Curr. Opin. Genet. Dev. 2008, 18, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–37. [Google Scholar] [CrossRef] [PubMed]
- Skobe, M.; Hawighorst, T.; Jackson, D.G.; Prevo, R.; Janes, L.; Velasco, P.; Riccardi, L.; Alitalo, K.; Claffey, K.; Detmar, M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 2001, 7, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, J.P.; Thiele, W. Tumor metastasis and the lymphatic vasculature. Int. J. Cancer 2009, 125, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Ran, S.; Volk, L.; Hall, K.; Flister, M.J. Lymphangiogenesis and lymphatic metastasis in breast cancer. Pathophysiology 2010, 17, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Nisato, R.E.; Tille, J.-C.; Pepper, M.S. Lymphangiogenesis and tumor metastasis. Thromb. Haemost. 2003, 90, 591–597. [Google Scholar] [PubMed]
- Tobler, N.E.; Detmar, M. Tumor and lymph node lymphangiogenesis—Impact on cancer metastasis. J. Leukoc. Biol. 2006, 80, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Thiele, W.; Sleeman, J.P. Tumor-induced lymphangiogenesis: A target for cancer therapy? J. Biotechnol. 2006, 124, 224–241. [Google Scholar]
- Wissmann, C.; Detmar, M. Pathways targeting tumor lymphangiogenesis. Clin. Cancer Res. 2006, 12, 6865–6868. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kozaki, K.-I.; Karpanen, T.; Koshikawa, K.; Yla-Herttuala, S.; Takahashi, T.; Alitalo, K. Suppression of tumor lymphangiogenesis and lymph node metastasis by blocking vascular endothelial growth factor receptor 3 signaling. J. Natl. Cancer Inst. 2002, 94, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Achen, M.G.; Stacker, S.A. Molecular control of lymphatic metastasis. Ann. N. Y. Acad. Sci. 2008, 1131, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, S.D. Insights into the mechanisms of lymph node metastasis. Cancer 2003, 98, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Wu, X.; Takekoshi, T.; Sullivan, A.; Hwang, S.T. Inflammation and tumor microenvironment in lymph node metastasis. Cancers 2011, 3, 927–944. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Schönbein, G.W. Microlymphatics and lymph flow. Physiol. Rev. 1990, 70, 987–1028. [Google Scholar]
- Swartz, M.A. The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 2001, 50, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.; Tunheim, S.; Mathiesen, B.; Graff, B.; Halsor, E.; Nilsen, K.; Galappathi, K. Pulmonary and lymph node metastasis is associated with primary tumor interstitial fluid pressure in human melanoma xenografts. Cancer Res. 2002, 62, 661–664. [Google Scholar] [PubMed]
- Hompland, T.; Ellingsen, C.; Øvrebø, K.M.; Rofstad, E.K. Interstitial fluid pressure and associated lymph node metastasis revealed in tumors by dynamic contrast-enhanced MRI. Cancer Res. 2012, 72, 4899–4908. [Google Scholar] [CrossRef]
- Moussion, C.; Girard, J.-P. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature 2011, 479, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Mionnet, C.; Sanos, S.L.; Mondor, I.; Jorquera, A.; Laugier, J.-P.; Germain, R.N.; Bajenoff, M. High endothelial venules as traffic control points maintaining lymphocyte population homeostasis in lymph nodes. Blood 2011, 118, 6115–6122. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Marshall, J.R.; King, M.R. Glycomechanics of the metastatic cascade: Tumor cell-endothelial cell interactions in the circulation. Ann. Biomed. Eng. 2012, 40, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Sher, B.T.; Bargatze, R.; Holzmann, B.; Gallatin, W.M.; Matthews, D.; Wu, N.; Picker, L.; Butcher, E.C.; Weissman, I.L. Homing receptors and metastasis. Adv. Cancer Res. 1988, 51, 361–390. [Google Scholar] [PubMed]
- Bargatze, R.F.; Wu, N.W.; Weissman, I.L.; Butcher, E.C. High endothelial venule binding as a predictor of the dissemination of passaged murine lymphomas. J. Exp. Med. 1987, 166, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Ozcinar, B.; Muslumanoglu, M.; Igci, A.; Gurdal, S.O.; Yavuz, E.; Kecer, M.; Dagoglu, T.; Ozmen, V. Clinical importance of micrometastasis in sentinel lymph nodes. Breast 2011, 20, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Mehes, G.; Witt, A.; Kubista, E.; Ambros, P.F. Classification of isolated tumor cells and micrometastasis. Cancer 2000, 89, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Noura, S.; Yamamoto, H.; Miyake, Y.; Kim, B.N.; Takayama, O.; Seshimo, I.; Ikenaga, M.; Ikeda, M.; Sekimoto, M.; Matsuura, N.; et al. Immunohistochemical assessment of localization and frequency of micrometastases in lymph nodes of colorectal cancer. Clin. Cancer Res. 2002, 8, 759–767. [Google Scholar] [PubMed]
- Weaver, D.L.; Krag, D.N.; Manna, E.A.; Ashikaga, T.; Waters, B.L.; Harlow, S.P.; Bauer, K.D.; Julian, T.B. Detection of occult sentinel lymph node micrometastases by immunohistochemistry in breast cancer. An NSABP protocol B-32 quality assurance study. Cancer 2006, 107, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Gerber, B.; Krause, A.; Muller, H.; Richter, D.; Reimer, T.; Makovitzky, J.; Herrnring, C.; Jeschke, U.; Kundt, G.; Friese, K. Simultaneous immunohistochemical detection of tumor cells in lymph nodes and bone marrow aspirates in breast cancer and its correlation with other prognostic factors. J. Clin. Oncol. 2001, 19, 960–971. [Google Scholar] [PubMed]
- Mori, M.; Mimori, K.; Inoue, H.; Barnard, G.F.; Tsuji, K.; Nanbara, S.; Ueo, H.; Akiyoshi, T. Detection of cancer micrometastases in lymph nodes by reverse transcriptase-polymerase chain reaction. Cancer Res. 1995, 55, 3417–3420. [Google Scholar] [PubMed]
- Slade, M.J.; Smith, B.M.; Sinnett, H.D.; Cross, N.C.; Coombes, R.C. Quantitative polymerase chain reaction for the detection of micrometastases in patients with breast cancer. J. Clin. Oncol. 1999, 17, 870–879. [Google Scholar] [PubMed]
- Sahai, E. Illuminating the metastatic process. Nat. Rev. Cancer 2007, 7, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Progress in human tumour immunology and immunotherapy. Nature 2001, 411, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. The tumor-draining lymph node as an immune-privileged site. Immunol. Rev. 2006, 213, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Cochran, A.J.; Huang, R.-R.; Morton, D.L.; Maecker, H.T. Immune responses in the draining lymph nodes against cancer: Implications for immunotherapy. Cancer Metastasis Rev. 2006, 25, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.F.; Arens, R.; Melief, C.J.M. Local targets for immune therapy to cancer: Tumor draining lymph nodes and tumor microenvironment. Int. J. Cancer 2013, 132, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Jeanbart, L.; Ballester, M.; de Titta, A.; Corthesy, P.; Romero, P.; Hubbell, J.A.; Swartz, M.A. Enhancing efficacy of anti-cancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol. Res. 2014. [Google Scholar] [CrossRef]
- Thomas, S.N.; Vokali, E.; Lund, A.W.; Hubbell, J.A.; Swartz, M.A. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 2014, 35, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Sixt, M.; Kanazawa, N.; Selg, M.; Samson, T.; Roos, G.; Reinhardt, D.P.; Pabst, R.; Lutz, M.B.; Sorokin, L. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 2005, 22, 19–29. [Google Scholar] [CrossRef]
- Roozendaal, R.; Mebius, R.E.; Kraal, G. The conduit system of the lymph node. Int. Immunol. 2008, 20, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Margolin, K. Tumor-infiltrating lymphocytes in melanoma. Curr. Oncol. Rep. 2012, 14, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Fu, Y.-X. Tumor-infiltrating T lymphocytes: Friends or foes? Lab. Investig. 2006, 86, 231–245. [Google Scholar] [CrossRef]
- Lança, T.; Silva-Santos, B. The split nature of tumor-infiltrating leukocytes: Implications for cancer surveillance and immunotherapy. Oncoimmunology 2012, 1, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Y.; Looi, K.S.; Tan, E.M. Identification of tumor-associated antigens as diagnostic and predictive biomarkers in cancer. Methods Mol. Biol. 2009, 520, 1–10. [Google Scholar]
- Pardoll, D. T cells take aim at cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 15840–15842. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lanier, L.L. Natural killer cells and cancer. Adv. Cancer Res. 2003, 90, 127–156. [Google Scholar] [PubMed]
- Cheent, K.; Khakoo, S.I. Natural killer cells: Integrating diversity with function. Immunology 2009, 126, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S.; Ballas, Z.K. Natural killer cells in human health and disease. Clin. Immunol. 2006, 118, 1–10. [Google Scholar] [CrossRef]
- Koch, J.; Steinle, A.; Watzl, C.; Mandelboim, O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013, 34, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural Killer (NK) cell–mediated cytotoxicity: Differential use of TRAIL and fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Aptsiauri, N.; Cabrera, T.; Mendez, R.; Garcia-Lora, A.; Ruiz-Cabello, F.; Garrido, F. Role of altered expression of HLA class I molecules in cancer progression. Adv. Exp. Med. Biol. 2007, 601, 123–131. [Google Scholar] [PubMed]
- Smyth, M.J.; Crowe, N.Y.; Godfrey, D.I. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int. Immunol. 2001, 13, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Takeda, K.; Wiltrout, R.H.; Sedger, L.M.; Kayagaki, N.; Yagita, H.; Okumura, K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001, 193, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.A.; Ley, K. Trafficking of natural killer cells. Curr. Mol. Med. 2004, 4, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.M.; Roberti, M.P.; Mordoh, J. Natural killer cells in human cancer: From biological functions to clinical applications. J. Biomed. Biotechnol. 2011, 2011, 676198. [Google Scholar] [CrossRef]
- Martín-Fontecha, A.; Thomsen, L.L.; Brett, S.; Gerard, C.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 2004, 5, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, G.; Thomas, D.; Lin, S.-L.; Goodman, K.; Morandi, B.; Muller, W.A.; Moretta, A.; Münz, C. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J. Immunol. 2004, 172, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Garrod, K.R.; Wei, S.H.; Parker, I.; Cahalan, M.D. Natural killer cells actively patrol peripheral lymph nodes forming stable conjugates to eliminate MHC-mismatched targets. Proc. Natl. Acad. Sci. USA 2007, 104, 12081–12086. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: A potential new link between adaptive and innate immunity. Blood 2003, 101, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Bajénoff, M.; Breart, B.; Huang, A.Y.C.; Qi, H.; Cazareth, J.; Braud, V.M.; Germain, R.N.; Glaichenhaus, N. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J. Exp. Med. 2006, 203, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Messaoudene, M.; Fregni, G.; Fourmentraux-Neves, E.; Chanal, J.; Maubec, E.; Mazouz-Dorval, S.; Couturaud, B.; Girod, A.; Sastre-Garau, X.; Albert, S.; et al. Ge mature cytotoxic CD56bright/CD16+ natural killer cells can infiltrate lymph nodes adjacent to metastatic melanoma. Cancer Res. 2014, 74, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Baskic, D.; Vujanovic, L.; Arsenijevic, N.; Whiteside, T.L.; Myers, E.N.; Vujanovic, N.L. Suppression of natural killer-cell and dendritic-cell apoptotic tumoricidal activity in patients with head and neck cancer. Head Neck 2013, 35, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Mamessier, E.; Sylvain, A.; Thibult, M.-L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Gonçalves, A.; André, P.; Romagné, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef]
- Doubrovina, E.S.; Doubrovin, M.M.; Vider, E.; Sisson, R.B.; O’Reilly, R.J.; Dupont, B.; Vyas, Y.M. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J. Immunol. 2003, 171, 6891–6899. [Google Scholar] [CrossRef] [PubMed]
- Hersey, P.; Zhang, X.D. How melanoma cells evade trail-induced apoptosis. Nat. Rev. Cancer 2001, 1, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Van Mierlo, G.J.D.; Boonman, Z.F.H.M.; Dumortier, H.M.H.; den Boer, A.T.; Fransen, M.F.; Nouta, J.; van der Voort, E.I.H.; Offringa, R.; Toes, R.E.M.; Melief, C.J.M. Activation of dendritic cells that cross-present tumor-derived antigen licenses CD8+ CTL to cause tumor eradication. J. Immunol. 2004, 173, 6753–6759. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.J.; Boldajipour, B.; Beemiller, P.; Pandurangi, P.; Sorensen, C.; Werb, Z.; Egeblad, M.; Krummel, M.F. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell 2012, 21, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Spiotto, M.T.; Lee, Y.; Schreiber, H.; Fu, Y.-X. Complementary role of CD4+ T cells and secondary lymphoid tissues for cross-presentation of tumor antigen to CD8+ T cells. J. Exp. Med. 2003, 197, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Nagorsen, D.; Scheibenbogen, C.; Marincola, F.M.; Letsch, A.; Keilholz, U. Natural T cell immunity against cancer. Clin. Cancer Res. 2003, 9, 4296–4303. [Google Scholar]
- Petersen, T.R.; Dickgreber, N.; Hermans, I.F. Tumor antigen presentation by dendritic cells. Crit. Rev. Immunol. 2010, 30, 345–386. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.; Hayashi, R.; Lafond-Walker, A.; Lowenstein, C.; Pardoll, D.; Levitsky, H. The central role of CD4(+) T cells in the antitumor immune response. J. Exp. Med. 1998, 188, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A.; Skovseth, D.K.; Lundin, K.U.; Røsjø, E.; Omholt, H.; Hofgaard, P.O.; Haraldsen, G.; Bogen, B. Primary antitumor immune response mediated by CD4+ T cells. Immunity 2005, 22, 371–383. [Google Scholar] [CrossRef]
- Rizzuto, G.A.; Merghoub, T.; Hirschhorn-Cymerman, D.; Liu, C.; Lesokhin, A.M.; Sahawneh, D.; Zhong, H.; Panageas, K.S.; Perales, M.-A.; Altan-Bonnet, G.; et al. Self-antigen-specific CD8+ T cell precursor frequency determines the quality of the antitumor immune response. J. Exp. Med. 2009, 206, 849–866. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Kitayama, J.; Muto, T.; Nagawa, H. Characterization of intracellular cytokine profile of CD4(+) T cells in peripheral blood and tumor-draining lymph nodes of patients with gastrointestinal cancer. Jpn. J. Clin. Oncol. 2000, 30, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Torisu-Itakara, H.; Cochran, A.J.; Kadison, A.; Huynh, Y.; Morton, D.L.; Essner, R. Quantitative analysis of melanoma-induced cytokine-mediated immunosuppression in melanoma sentinel nodes. Clin. Cancer Res. 2005, 11, 107–112. [Google Scholar] [PubMed]
- Yang, Y.; Huang, C.-T.; Huang, X.; Pardoll, D.M. Persistent toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat. Immunol. 2004, 5, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.F. CD8+ regulatory T cells, their suppressive mechanisms, and regulation in cancer. Hum. Immunol. 2008, 69, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Kim, J.H.; Falo, L.D.; You, Z. Tumor regulatory T cells potently abrogate antitumor immunity. J. Immunol. 2009, 182, 6160–6167. [Google Scholar] [CrossRef] [PubMed]
- James, E.; Yeh, A.; King, C.; Korangy, F.; Bailey, I.; Boulanger, D.S.; van den Eynde, B.J.; Murray, N.; Elliott, T.J. Differential suppression of tumor-specific CD8+ T cells by regulatory T cells. J. Immunol. 2010, 185, 5048–5055. [Google Scholar] [CrossRef] [PubMed]
- Grauer, O.M.; Nierkens, S.; Bennink, E.; Toonen, L.W.J.; Boon, L.; Wesseling, P.; Sutmuller, R.P.M.; Adema, G.J. CD4+ FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int. J. Cancer 2007, 121, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Kiniwa, Y.; Miyahara, Y.; Wang, H.Y.; Peng, W.; Peng, G.; Wheeler, T.M.; Thompson, T.C.; Old, L.J.; Wang, R.-F. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin. Cancer Res. 2007, 13, 6947–6958. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Viehl, C.T.; Moore, T.T.; Liyanage, U.K.; Frey, D.M.; Ehlers, J.P.; Eberlein, T.J.; Goedegebuure, P.S.; Linehan, D.C. Depletion of CD4+ CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann. Surg. Oncol. 2006, 13, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Dercamp, C.; Chemin, K.; Caux, C.; Trinchieri, G.; Vicari, A.P. Distinct and overlapping roles of interleukin-10 and CD25+ regulatory T cells in the inhibition of antitumor CD8 T-cell responses. Cancer Res. 2005, 65, 8479–8486. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Kjaergäard, J.; Plautz, G.E.; Awad, M.; Drazba, J.A.; Shu, S.; Cohen, P.A. Tumor-induced L-selectin high suppressor T cells mediate potent effector T cell blockade and cause failure of otherwise curative adoptive immunotherapy. J. Immunol. 2002, 169, 4811–4821. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H.G. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Wolchok, J.D. Tumor antigens for cancer immunotherapy: Therapeutic potential of xenogeneic DNA vaccines. J. Transl. Med. 2004, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, A.M.; Robinson, B.W.S.; Currie, A.J. Tumor antigen cross-presentation and the dendritic cell: Where it all begins? Clin. Dev. Immunol. 2010, 2010, 539519. [Google Scholar]
- Joshi, M.D.; Unger, W.J.; Storm, G.; van Kooyk, Y.; Mastrobattista, E. Targeting tumor antigens to dendritic cells using particulate carriers. J. Control. Release 2012, 161, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.J.; Morton, D.L.; Stern, S.; Lana, A.M.; Essner, R.; Wen, D.R. Sentinel lymph nodes show profound downregulation of antigen-presenting cells of the paracortex: Implications for tumor biology and treatment. Mod. Pathol. 2001, 14, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Almand, B.; Resser, J.R.; Lindman, B.; Nadaf, S.; Clark, J.I.; Kwon, E.D.; Carbone, D.P.; Gabrilovich, D.I. Clinical significance of defective dendritic cell differentiation in cancer. Clin. Cancer Res. 2000, 6, 1755–1766. [Google Scholar] [PubMed]
- Harris, N.L.; Ronchese, F. The role of B7 costimulation in T-cell immunity. Immunol. Cell Biol. 1999, 77, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.S.; Flavell, R.A. The role of CD40 ligand in costimulation and T-cell activation. Immunol. Rev. 1996, 153, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Shuford, W.W.; Klussman, K.; Tritchler, D.D.; Loo, D.T.; Chalupny, J.; Siadak, A.W.; Brown, T.J.; Emswiler, J.; Raecho, H.; Larsen, C.P.; et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med. 1997, 186, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Scholer, A.; Hugues, S.; Boissonnas, A.; Fetler, L.; Amigorena, S. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity 2008, 28, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M. Tumor-altered dendritic cell function: Implications for anti-tumor immunity. Front. Immunol. 2013, 4, 192. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.-Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.J.; Huang, R.-R.; Itakura, E.; Lee, J.H.; Molenkamp, B.G. Dendritic Cells in Tumor-Draining Lymph Nodes; Salter, R.D., Shurin, M.R., Eds.; Springer: New York, NY, USA, 2009; pp. 291–307. [Google Scholar]
- Fremd, C.; Schuetz, F.; Sohn, C.; Beckhove, P.; Domschke, C. B cell-regulated immune responses in tumor models and cancer patients. Oncoimmunology 2013, 2, e25443. [Google Scholar] [CrossRef] [PubMed]
- Coronella-Wood, J.A.; Hersh, E.M. Naturally occurring B-cell responses to breast cancer. Cancer Immunol. Immunother. 2003, 52, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Grover, A.C.; Donald, E.J.; Carr, A.; Yu, J.; Whitfield, J.; Nelson, M.; Takeshita, N.; Chang, A.E. Simultaneous targeting of CD3 on T cells and CD40 on B or dendritic cells augments the antitumor reactivity of tumor-primed lymph node cells. J. Immunol. 2005, 175, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Marits, P.; Zirakzadeh, A.A.; Sherif, A.; Winqvist, O. The many flavors of tumor-associated B cells. Oncoimmunology 2013, 2, e25237. [Google Scholar] [CrossRef] [PubMed]
- Zirakzadeh, A.A.; Marits, P.; Sherif, A.; Winqvist, O. Multiplex B cell characterization in blood, lymph nodes, and tumors from patients with malignancies. J. Immunol. 2013, 190, 5847–5855. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Schlößer, H.A.; Stippel, D.L.; Theurich, S.; von Bergwelt-Baildon, M. Comment on “Multiplex B cell characterization in blood, lymph nodes, and tumors from patients with malignancies.”. J. Immunol. 2013, 191, 4471. [Google Scholar] [CrossRef] [PubMed]
- Marits, P.; Zirakzadeh, A.A.; Sherif, A.; Winqvist, O. Response to comment on “Multiplex B Cell Characterization in Blood, Lymph Nodes, and Tumors from Patients with Malignancies.”. J. Immunol. 2013, 191, 4471–4472. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveilance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K.; Arihiro, K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006, 66, 5527–5536. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; San Roman, S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, A.S.; Holtan, S.G.; Grotz, T.E.; Allred, J.B.; Jakub, J.W.; Erickson, L.A.; Markovic, S.N. Regional immunity in melanoma: Immunosuppressive changes precede nodal metastasis. Mod. Pathol. 2011, 24, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, N.S.; Yu, H.; Simons, D.L.; Bhattacharya, N.; Carcamo-Cavazos, V.; Yan, N.; Dirbas, F.M.; Johnson, D.L.; Schwartz, E.J.; Lee, P.P. Altered local and systemic immune profiles underlie lymph node metastasis in breast cancer patients. Int. J. Cancer 2013, 132, 2537–2547. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Palucka, A.K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005, 5, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Sabado, R.L.; Bhardwaj, N. Directing dendritic cell immunotherapy towards successful cancer treatment. Immunotherapy 2010, 2, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J.M. Cancer immunotherapy by dendritic cells. Immunity 2008, 29, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Schuler, G. Dendritic cells in cancer immunotherapy. Eur. J. Immunol. 2010, 40, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, J.E.; Bonehill, A.; Thielemans, K.; Wan, Y. Engineering dendritic cells to enhance cancer immunotherapy. Mol. Ther. 2011, 19, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.G.M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology 2009, 23, 488–496. [Google Scholar] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O.; Barth, N.M.; VanderMolen, L.A.; Mahdavi, K.; McClure, S.E. Should high-dose interleukin-2 still be the preferred treatment for patients with metastatic melanoma? Cancer Biother. Radiopharm. 2012, 27, 337–343. [Google Scholar]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Parmiani, G.; Castelli, C.; Dalerba, P.; Mortarini, R.; Rivoltini, L.; Marincola, F.M.; Anichini, A. Cancer immunotherapy with peptide-based vaccines: What have we achieved? Where are we going? J. Natl. Cancer Inst. 2002, 94, 805–818. [Google Scholar] [CrossRef]
- Kwon, E.D.; Hurwitz, A.A.; Foster, B.A.; Madias, C.; Feldhaus, A.L.; Greenberg, N.M.; Burg, M.B.; Allison, J.P. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 8099–8103. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.G.; Schultz, E.S. Dendritic cell-based immunotherapy. Curr. Top. Microbiol. Immunol. 2003, 276, 163–197. [Google Scholar] [PubMed]
- Fernandez, N.; Duffour, M.T.; Perricaudet, M.; Lotze, M.T.; Tursz, T.; Zitvogel, L. Active specific T-cell-based immunotherapy for cancer: Nucleic acids, peptides, whole native proteins, recombinant viruses, with dendritic cell adjuvants or whole tumor cell-based vaccines. Principles and future prospects. Cytokines Cell Mol. Ther. 1998, 4, 53–65. [Google Scholar] [PubMed]
- Paulis, L.E.; Mandal, S.; Kreutz, M.; Figdor, C.G. Dendritic cell-based nanovaccines for cancer immunotherapy. Curr. Opin. Immunol. 2013, 25, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Jesorka, A.; Orwar, O. Liposomes: Technologies and analytical applications. Annu. Rev. Anal. Chem. 2008, 1, 801–832. [Google Scholar] [CrossRef]
- Cˇeh, B.; Winterhalter, M.; Frederik, P.M.; Vallner, J.J.; Lasic, D.D. Stealth® liposomes: From theory to product. Adv. Drug Deliv. Rev. 1997, 24, 165–177. [Google Scholar] [CrossRef]
- Oussoren, C.; Storm, G. Liposomes to target the lymphatics by subcutaneous administration. Adv. Drug Deliv. Rev. 2001, 50, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Phillips, W.T.; Goins, B.A.; Medina, L.A. Targeting of liposomes to lymph nodes. In Liposome Technology; Informa Healthcare: New York, NY, USA, 2006; Volume 3, pp. 13–231. [Google Scholar]
- Altin, J.G.; Parish, C.R. Liposomal vaccines-targeting the delivery of antigen. Methods 2006, 40, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Fleiner, M.; Benzinger, P.; Fichert, T.; Massing, U. Studies on protein−liposome coupling using novel thiol-reactive coupling lipids: Influence of spacer length and polarity. Bioconjug. Chem. 2001, 12, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Van Broekhoven, C.L.; Altin, J.G. A novel system for convenient detection of low-affinity receptor-ligand interactions: Chelator-lipid liposomes engrafted with recombinant CD4 bind to cells expressing MHC class II. Immunol. Cell Biol. 2001, 79, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Arigita, C.; Sundblad, A.; Jiskoot, W.; Storm, G.; Frokjaer, S. Interaction of dendritic cells with antigen-containing liposomes: Effect of bilayer composition. Vaccine 2004, 22, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Van Broekhoven, C.L.; Parish, C.R.; Demangel, C.; Britton, W.J.; Altin, J.G. Targeting dendritic cells with antigen-containing liposomes: A highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 2004, 64, 4357–4365. [Google Scholar] [CrossRef] [PubMed]
- Storni, T.; Kündig, T.M.; Senti, G.; Johansen, P. Immunity in response to particulate antigen-delivery systems. Adv. Drug Deliv. Rev. 2005, 57, 333–355. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, Y.; Niwa, T.; Biao, L.; Ikehara, S.K.; Ohashi, N.; Kobayashi, T.; Shimizu, Y.; Kojima, N.; Nakanishi, H. A carbohydrate recognition-based drug delivery and controlled release system using intraperitoneal macrophages as a cellular vehicle. Cancer Res. 2006, 66, 8740–8748. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, Y.; Shiuchi, N.; Kabata-Ikehara, S.; Nakanishi, H.; Yokoyama, N.; Takagi, H.; Nagata, T.; Koide, Y.; Kuzushima, K.; Takahashi, T.; et al. Effective induction of anti-tumor immune responses with oligomannose-coated liposome targeting to intraperitoneal phagocytic cells. Cancer Lett. 2008, 260, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Kojima, N.; Biao, L.; Nakayama, T.; Ishii, M.; Ikehara, Y.; Tsujimura, K. Oligomannose-coated liposomes as a therapeutic antigen-delivery and an adjuvant vehicle for induction of in vivo tumor immunity. J. Control. Release 2008, 129, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Kojima, N.; Ishii, M.; Kawauchi, Y.; Takagi, H. Oligomannose-coated liposome as a novel adjuvant for the induction of cellular immune responses to control disease status. Biomed. Res. Int. 2013, 2013, 562924. [Google Scholar] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- French, R.R.; Chan, H.T.; Tutt, A.L.; Glennie, M.J. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat. Med. 1999, 5, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.L.; O’Brien, L.; Hussain, A.; Crowther, G.R.; French, R.R.; Glennie, M.J. T cell immunity to lymphoma following treatment with anti-CD40 monoclonal antibody. J. Immunol. 2002, 168, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Flaherty, K.T.; Khalil, M.; Stumacher, M.S.; Bajor, D.L.; Hutnick, N.A.; Sullivan, P.; Mahany, J.J.; Gallagher, M.; Kramer, A.; et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J. Clin. Oncol. 2007, 25, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Development of TLR9 agonists for cancer therapy. J. Clin. Investig. 2007, 117, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Heikenwalder, M.; Polymenidou, M.; Junt, T.; Sigurdson, C.; Wagner, H.; Akira, S.; Zinkernagel, R.; Aguzzi, A. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat. Med. 2004, 10, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Kwong, B.; Liu, H.; Irvine, D.J. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials 2011, 32, 5134–5147. [Google Scholar] [CrossRef] [PubMed]
- Karanth, H.; Murthy, R.S.R. pH-sensitive liposomes—Principle and application in cancer therapy. J. Pharm. Pharmacol. 2007, 59, 469–483. [Google Scholar] [CrossRef]
- Yuba, E.; Harada, A.; Sakanishi, Y.; Watarai, S.; Kono, K. A liposome-based antigen delivery system using pH-sensitive fusogenic polymers for cancer immunotherapy. Biomaterials 2013, 34, 3042–3052. [Google Scholar] [CrossRef] [PubMed]
- Yuba, E.; Tajima, N.; Yoshizaki, Y.; Harada, A.; Hayashi, H.; Kono, K. Dextran derivative-based pH-sensitive liposomes for cancer immunotherapy. Biomaterials 2014, 35, 3091–3101. [Google Scholar] [CrossRef] [PubMed]
- Leitner, J.; Grabmeier-Pfistershammer, K.; Steinberger, P. Receptors and ligands implicated in human T cell costimulatory processes. Immunol. Lett. 2010, 128, 89–97. [Google Scholar] [CrossRef]
- Leitner, J.; Kuschei, W.; Grabmeier-Pfistershammer, K.; Woitek, R.; Kriehuber, E.; Majdic, O.; Zlabinger, G.; Pickl, W.F.; Steinberger, P. T cell stimulator cells, an efficient and versatile cellular system to assess the role of costimulatory ligands in the activation of human T cells. J. Immunol. Methods 2010, 362, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Kwong, B.; Gai, S.A.; Elkhader, J.; Wittrup, K.D.; Irvine, D.J. Localized Immunotherapy via liposome-anchored anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. 2013, 73, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Kwon, B.S. Immunotherapy of cancer with 4-1BB. Mol. Cancer Ther. 2012, 11, 1062–1070. [Google Scholar] [PubMed]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellström, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [PubMed]
- Phan, G.Q.; Attia, P.; Steinberg, S.M.; White, D.E.; Rosenberg, S.A. Factors associated with response to high-dose interleukin-2 in patients with metastatic melanoma. J. Clin. Oncol. 2001, 19, 3477–3482. [Google Scholar] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Stephan, M.T.; Gai, S.A.; Abraham, W.; Shearer, A.; Irvine, D.J. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J. Control. Release 2013, 172, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L.; Ponti, C.; Mirandola, P.; Gobbi, G.; Papa, S.; Galeotti, L.; Cocco, L.; Vitale, M. NK cells and cancer. J. Immunol. 2007, 178, 4011–4016. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; McGuire, M.J.; King, M.R. Sweeping lymph node micrometastases off their feet: An engineered model to evaluate natural killer cell mediated therapeutic intervention of circulating tumor cells that disseminate to the lymph nodes. Lab Chip 2014, 14, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Nguyen, C.B.; Kelley, S.K.; Dybdal, N.; Escand, E. Tissue distribution, stability, and pharmacokinetics of Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand in human colon carcinoma COLO205 tumor-bearing nude mice. Pharmacology 2004, 32, 1230–1238. [Google Scholar]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Wayne, E.; Rana, K.; Schaffer, C.B.; King, M.R. TRAIL-coated leukocytes that kill cancer cells in the circulation. Proc. Natl. Acad. Sci. USA 2014, 113, 930–935. [Google Scholar] [CrossRef]
- Muller, W.A. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003, 24, 326–333. [Google Scholar] [CrossRef]
- Zarbock, A.; Ley, K.; McEver, R.P.; Hidalgo, A. Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011, 118, 6743–6751. [Google Scholar] [PubMed]
- Mitchell, M.J.; King, M.R. Physical biology in cancer. 3. The role of cell glycocalyx in vascular transport of circulating tumor cells. Am. J. Physiol. Cell Physiol. 2014, 306, C89–C97. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekaran, S.; King, M.R. Microenvironment of Tumor-Draining Lymph Nodes: Opportunities for Liposome-Based Targeted Therapy. Int. J. Mol. Sci. 2014, 15, 20209-20239. https://doi.org/10.3390/ijms151120209
Chandrasekaran S, King MR. Microenvironment of Tumor-Draining Lymph Nodes: Opportunities for Liposome-Based Targeted Therapy. International Journal of Molecular Sciences. 2014; 15(11):20209-20239. https://doi.org/10.3390/ijms151120209
Chicago/Turabian StyleChandrasekaran, Siddarth, and Michael R. King. 2014. "Microenvironment of Tumor-Draining Lymph Nodes: Opportunities for Liposome-Based Targeted Therapy" International Journal of Molecular Sciences 15, no. 11: 20209-20239. https://doi.org/10.3390/ijms151120209