Multiple Mechanisms Mediate Resistance to Sorafenib in Urothelial Cancer

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
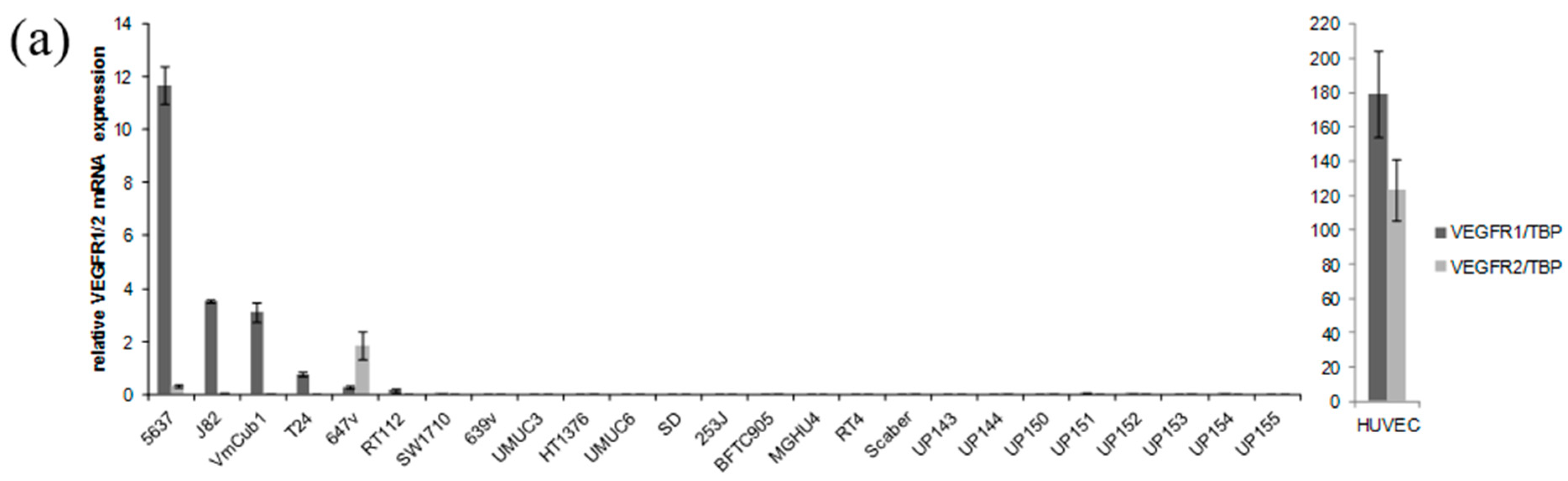
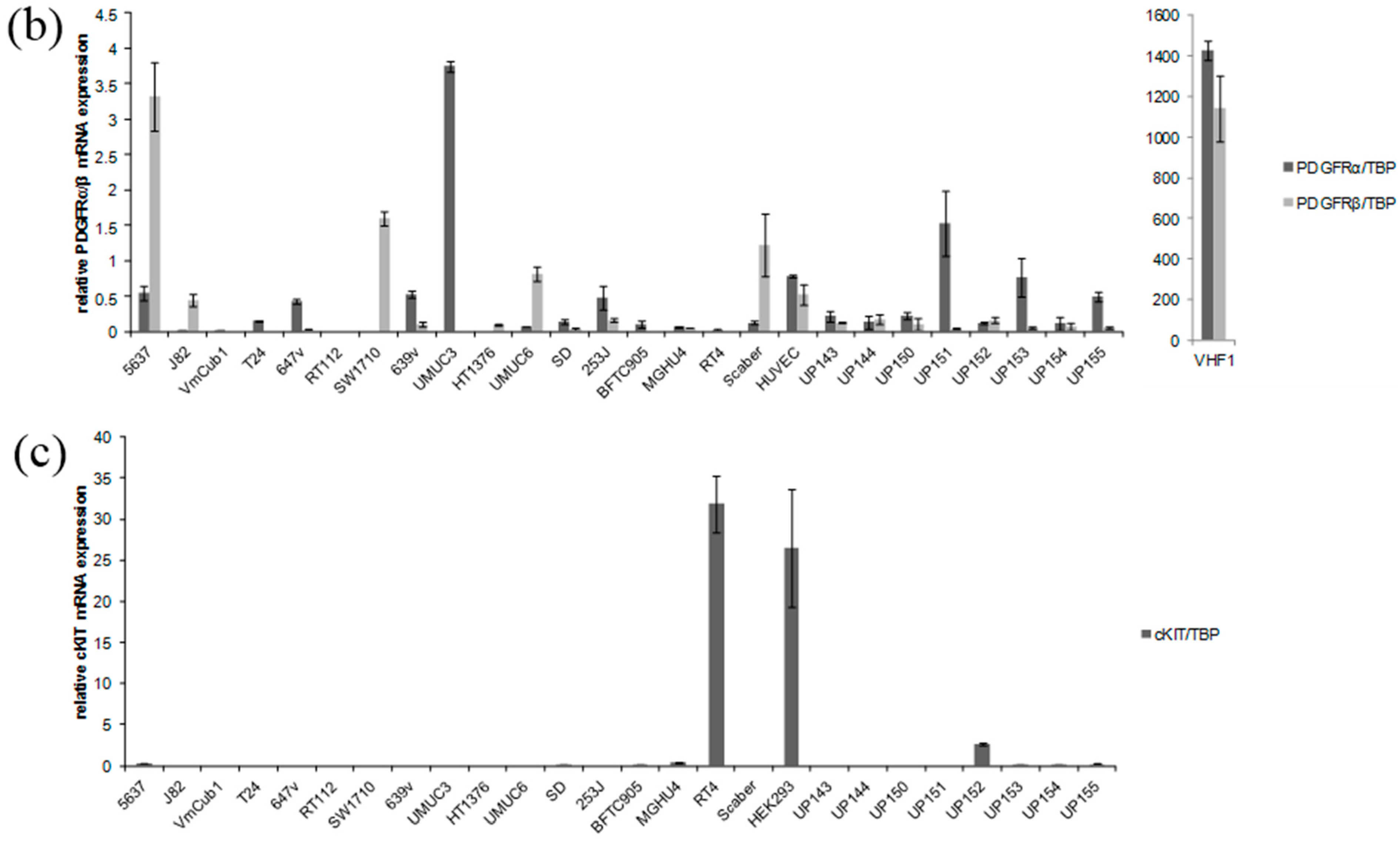
2.1. Receptor Tyrosine Kinases Targeted by Sorafenib Are Weakly Expressed in Urothelial Cancer Cell Lines (UCCs)


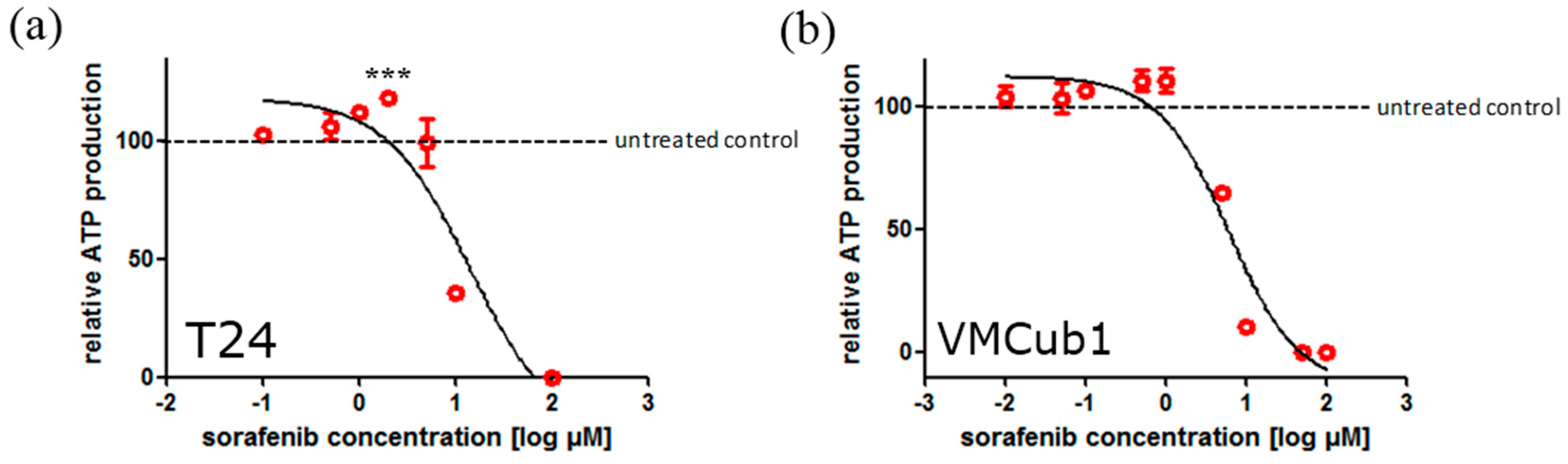
2.2. Sorafenib Treatment may Result in Both Increased and Decreased Cell Viability Depending on Dosage

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Sorafenib IC50 (µM) |
|---|---|
| T24 | 18.0 |
| 647v | 16.9 |
| SW1710 | 13.3 |
| VMCub1 | 8.4 |
| J82 | 8.1 |
| 5637 | 7.7 |
| UM-UC-3 | 6.7 |
| SD | 5.1 |
| 639v | 4.6 |
| UP199 | 1.6 |
| UP202 | 0.5 |

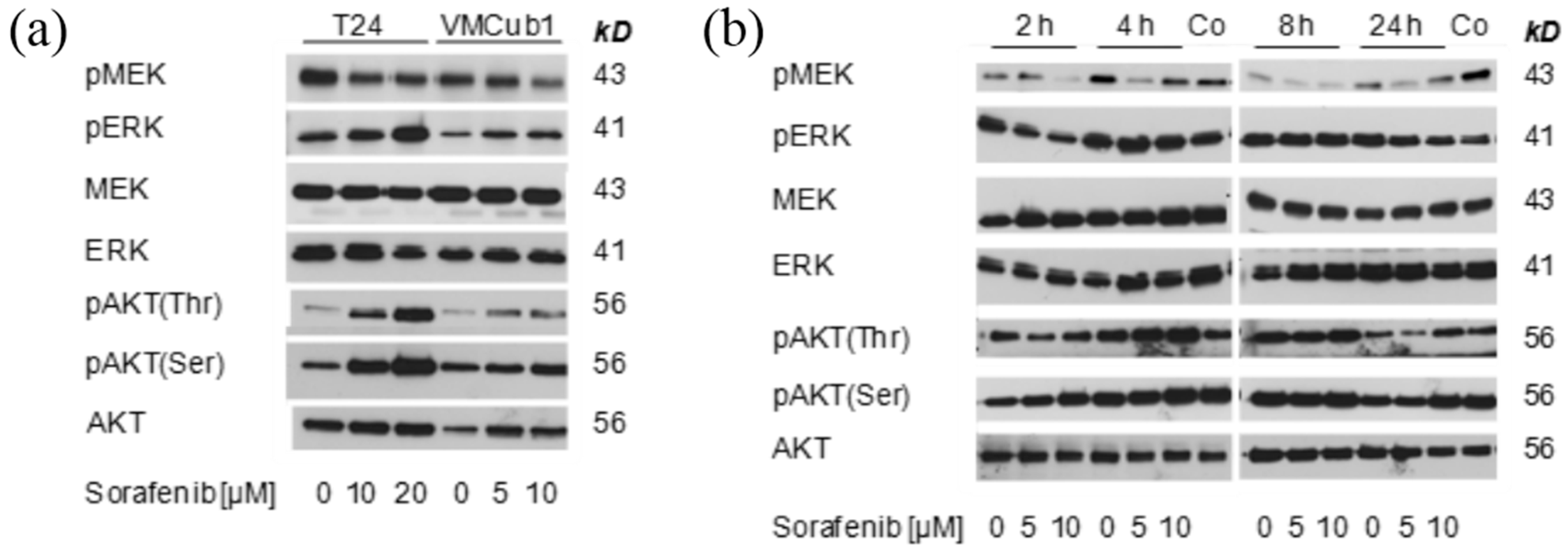
2.3. MAPK Signaling after Treatment with Sorafenib


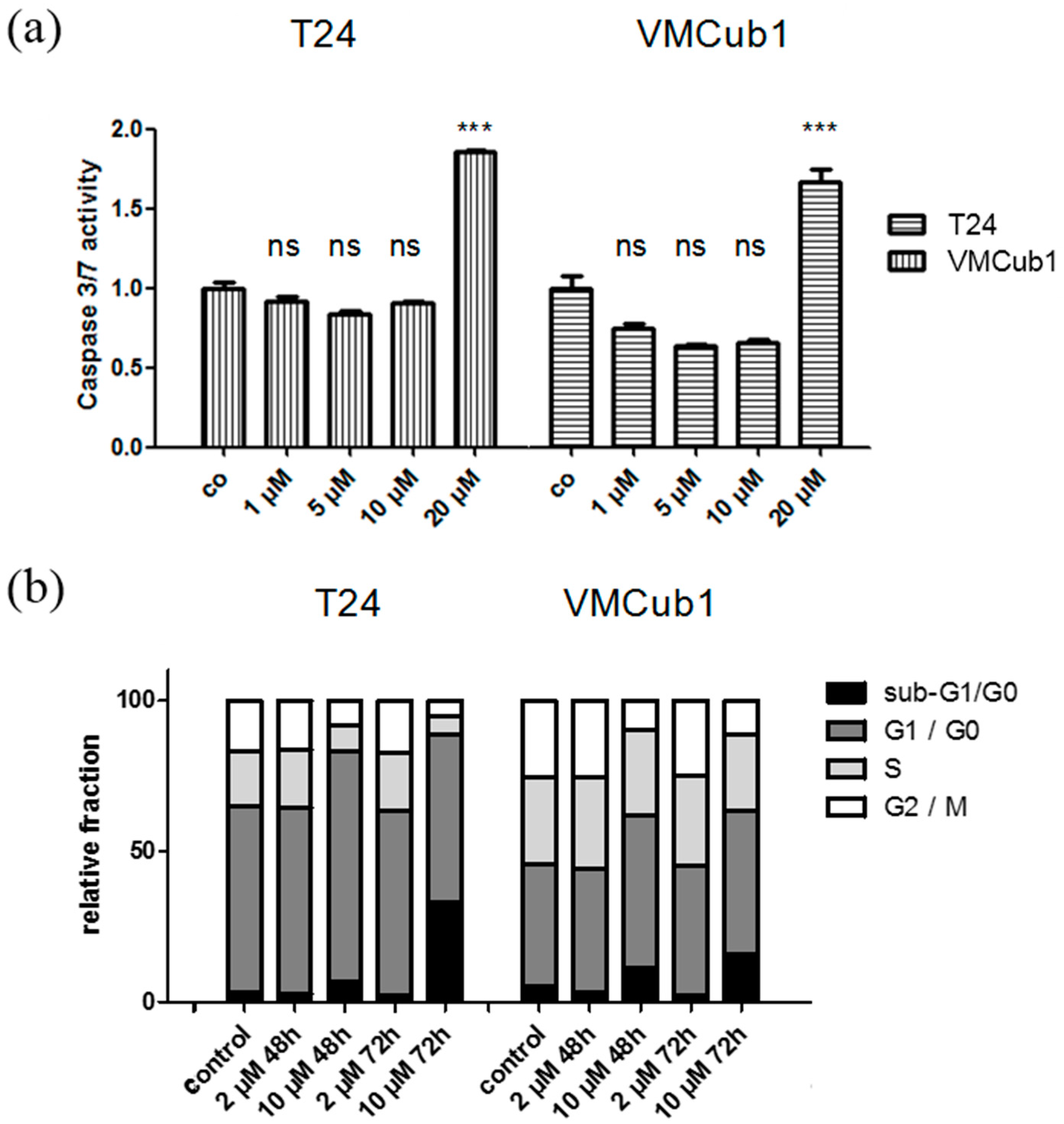
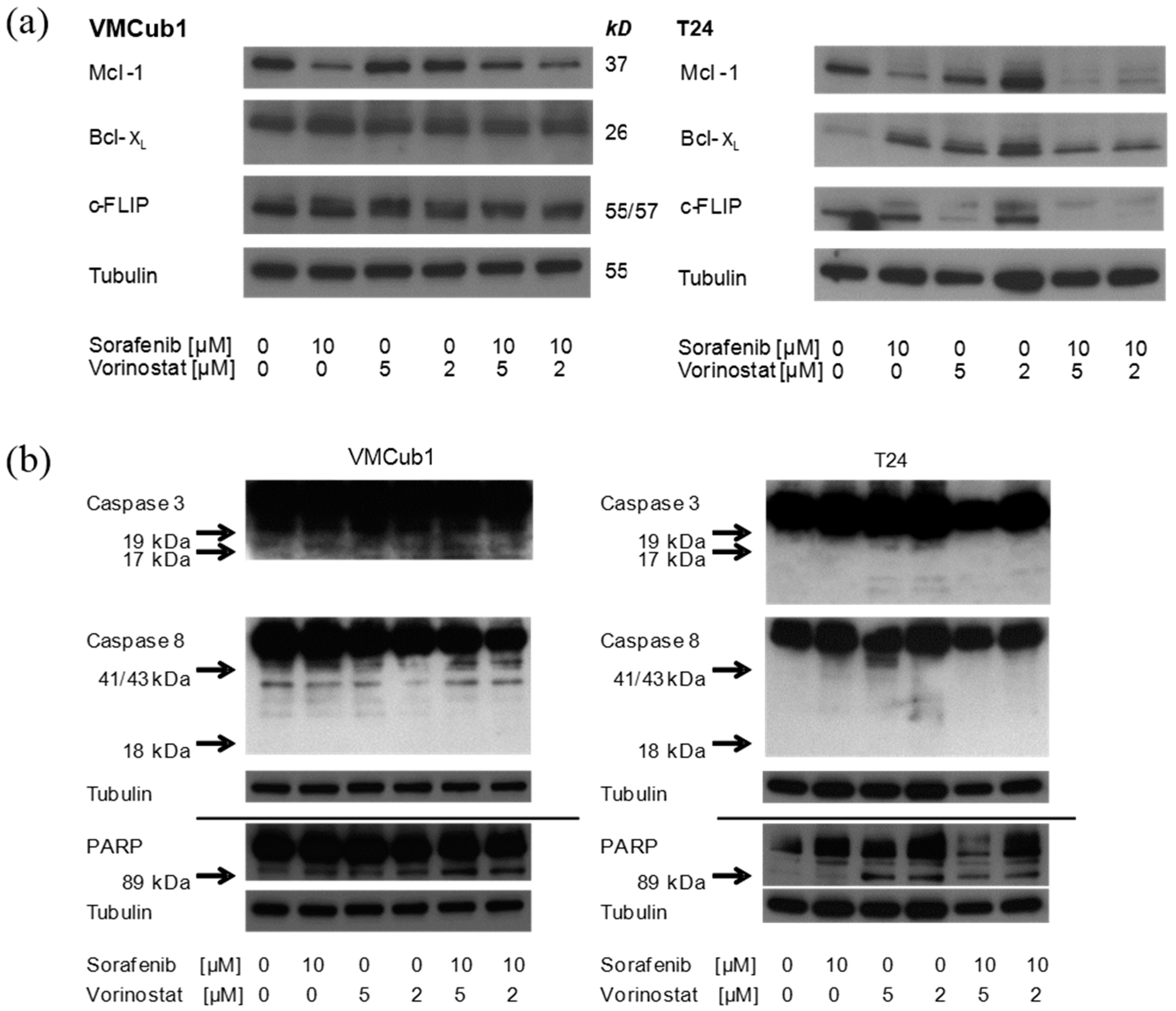
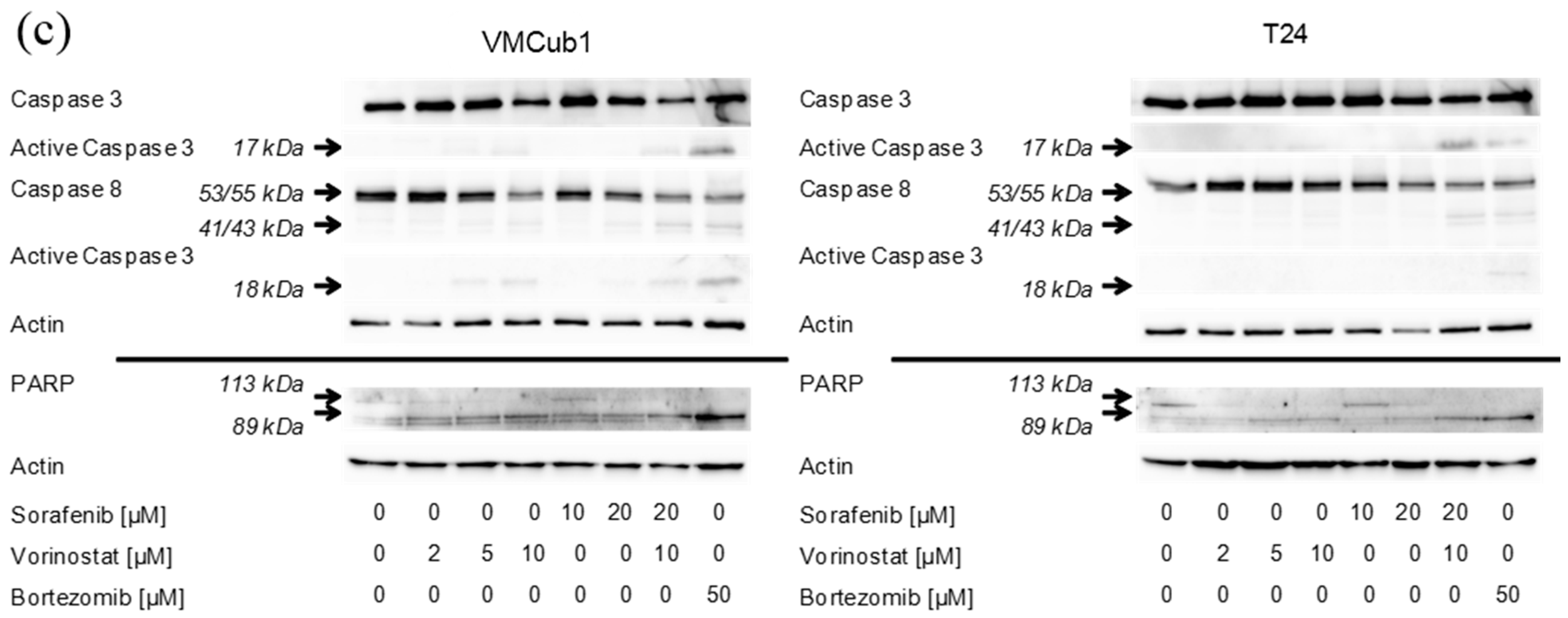
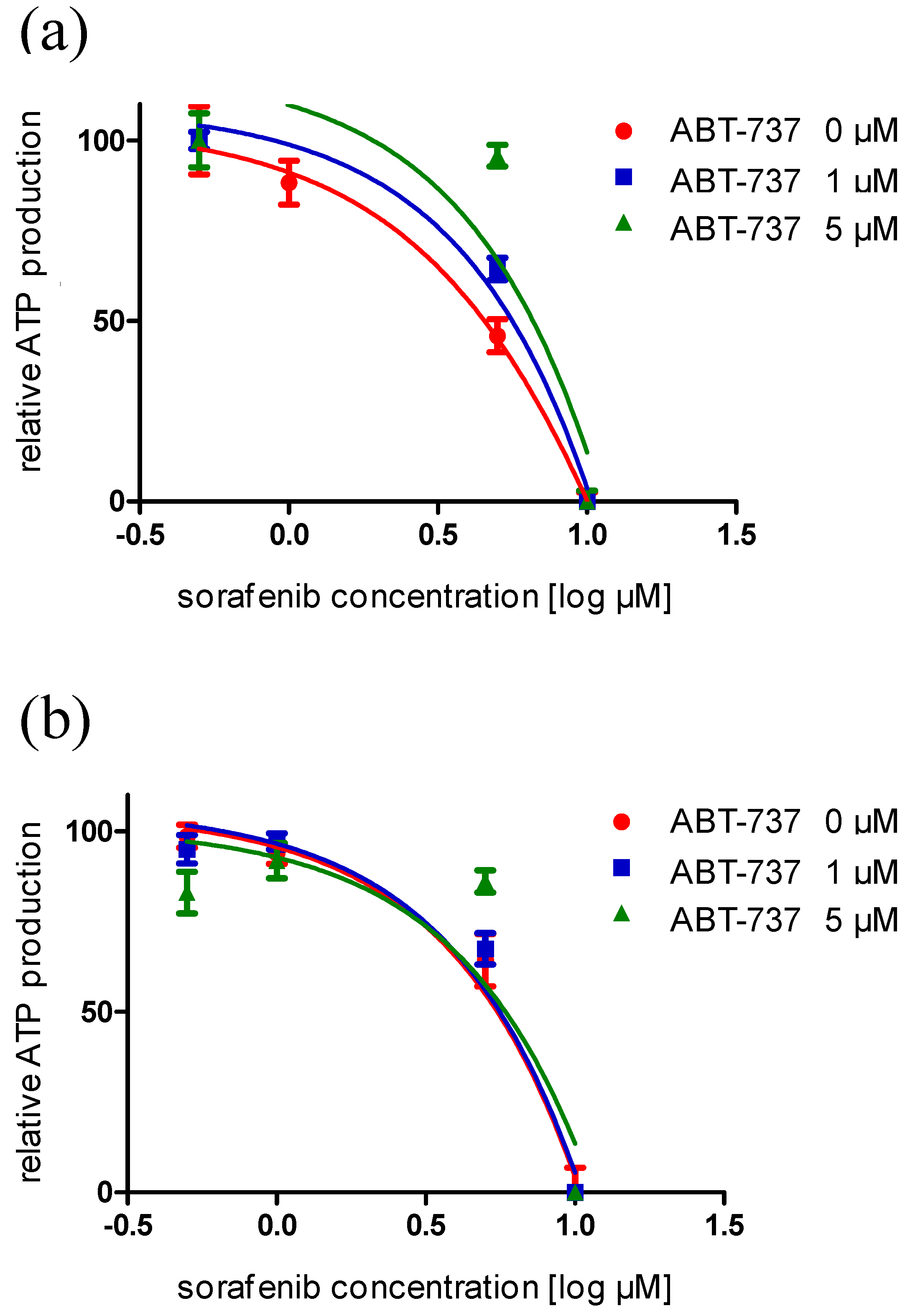
2.4. An Anti-Apoptotic State of UCCs Hampers Efficient Activation of Intrinsic Apoptosis


3. Experimental Section
3.1. Cell Lines and Cell Cultivation Procedures

3.2. Determination of Cell Viability, Mean IC50 and Caspase 3/7 Activity
3.3. Cell Cycle Analysis by Flow Cytometry
3.4. RNA Extraction and Analysis
3.5. Western Blot Analysis
3.6. Compounds
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Comperat, E.; Cowan, N.C.; de Santis, M.; Gakis, G.; Lebret, T.; Ribal, M.J.; van der Heijden, A.G.; Sherif, A. EAU guidelines on muscle-invasive and metastatic bladder cancer: Summary of the 2013 guidelines. Eur. Urol. 2013, 65, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.A. Understanding urothelial carcinoma through cancer pathways. Int. J. Cancer 2006, 119, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A. Bladder cancer subtypes defined by genomic alterations. Scand. J. Urol. Nephrol. Suppl. 2008, 42, 116–130. [Google Scholar] [CrossRef]
- Chow, N.H.; Liu, H.S.; Lee, E.I.C.; Chang, C.J.; Chan, S.H.; Cheng, H.L.; Tzai, T.S.; Lin, J.S.N. Significance of urinary epidermal growth factor and its receptor expression in human bladder cancer. Anticancer Res. 1997, 17, 1293–1296. [Google Scholar] [PubMed]
- Xia, G.; Kumar, S.R.; Hawes, D.; Cai, J.; Hassanieh, L.; Groshen, S.; Zhu, S.; Masood, R.; Quinn, D.I.; Broek, D.; et al. Expression and significance of vascular endothelial growth factor receptor 2 in bladder cancer. J. Urol. 2006, 175, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Chu, K.C.; Yeh, W.M. The expression of vascular endothelial growth factor in transitional cell carcinoma of urinary bladder is correlated with cancer progression. Urol. Oncol. 2004, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jebar, A.H.; Hurst, C.D.; Tomlinson, D.C.; Johnston, C.; Taylor, C.F.; Knowles, M.A. FGFR3 and RAS gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene 2005, 24, 5218–5225. [Google Scholar] [CrossRef] [PubMed]
- Otto, K.B.; Acharya, S.S.; Robinson, V.L. Stress-activated kinase pathway alteration is a frequent event in bladder cancer. Urol. Oncol. 2012, 30, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Available online: http://www.Ema.Europa.Eu/ema/index.Jsp?Curl=pages/medicines/human/medicines/000690/human_med_000929.Jsp&mid=wc0b01ac058001d124 (accessed on 28 october 2014).
- National Cancer Institute. Available online: http://www.Cancer.Gov/cancertopics/druginfo/fda-sorafenib-tosylate (accessed on 28 october 2014).
- Krege, S.; Rexer, H.; vom Dorp, F.; de Geeter, P.; Klotz, T.; Retz, M.; Heidenreich, A.; Kuhn, M.; Kamradt, J.; Feyerabend, S.; et al. Prospective randomized double-blind multicentre phase II study comparing gemcitabine and cisplatin plus sorafenib chemotherapy with gemcitabine and cisplatin plus placebo in locally advanced and/or metastasized urothelial cancer: Suse (AUO-AB 31/05). BJU Int. 2014, 113, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.S.; Winquist, E.; Eisen, A.; Hotte, S.J.; McWhirter, E.; Tannock, I.F.; Mukherjee, S.D.; Wang, L.S.; Blattler, C.; Wright, J.J.; et al. A phase II trial of sorafenib in first-line metastatic urothelial cancer: A study of the PMH phase II consortium. Investig. New Drug 2011, 29, 1045–1049. [Google Scholar] [CrossRef]
- Dreicer, R.; Li, H.L.; Stein, M.; di Paola, R.; Eleff, M.; Roth, B.J.; Wilding, G. Phase 2 trial of sorafenib in patients with advanced urothelial cancer a trial of the eastern cooperative oncology group. Cancer 2009, 115, 4090–4095. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.A.; Makonnen, S.; Lassoued, W.; Feldman, M.D.; Carter, C.; Lee, W.M. Inhibition of tumor endothelial ERK activation, angiogenesis, and tumor growth by sorafenib (BAY43-9006). Am. J. Pathol. 2006, 169, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Kopparapu, P.K.; Boorjian, S.A.; Robinson, B.D.; Downes, M.; Gudas, L.J.; Mongan, N.P.; Persson, J.L. Expression of VEGF and its receptors VEGFR1/VEGFR2 is associated with invasiveness of bladder cancer. Anticancer Res. 2013, 33, 2381–2390. [Google Scholar] [PubMed]
- Yang, S.; Wu, X.; Luo, C.; Pan, C.; Pu, J. Expression and clinical significance of hepacam and VEGF in urothelial carcinoma. World J. Urol. 2010, 28, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Went, P.T.; Dirnhofer, S.; Bundi, M.; Mirlacher, M.; Schraml, P.; Mangialaio, S.; Dimitrijevic, S.; Kononen, J.; Lugli, A.; Simon, R.; et al. Prevalence of KIT expression in human tumors. J. Clin. Oncol. 2004, 22, 4514–4522. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Leem, S.H.; Lee, S.Y.; Kim, S.C.; Park, E.S.; Kim, S.B.; Kim, S.K.; Kim, Y.J.; Kim, W.J.; Chu, I.S. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol. 2010, 28, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.; Grandoch, M.; vom Dorp, F.; Ruben, H.; Rosenkranz, A.; Fischer, J.W.; Weber, A.A. Stimulatory effects of the multi-kinase inhibitor sorafenib on human bladder cancer cells. Br. J. Pharmacol. 2010, 160, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. The RAF inhibitor paradox: Unexpected consequences of targeted drugs. Cancer Cell 2010, 17, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Swiatkowski, S.; Seifert, H.H.; Steinhoff, C.; Prior, A.; Thievessen, I.; Schliess, F.; Schulz, W.A. Activities of MAP-kinase pathways in normal uroepithelial cells and urothelial carcinoma cell lines. Exp. Cell Res. 2003, 282, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Varley, C.; Hill, G.; Pellegrin, S.; Shaw, N.J.; Selby, P.J.; Trejdosiewicz, L.K.; Southgate, J. Autocrine regulation of human urothelial cell proliferation and migration during regenerative responses in vitro. Exp. Cell Res. 2005, 306, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Leite, R.; Carreira, I.; Melo, J.; Ferreira, S.I.; Ribeiro, I.; Ferreira, J.; Filipe, M.; Bernardo, C.; Arantes-Rodrigues, R.; Oliveira, P.; et al. Genomic characterization of three urinary bladder cancer cell lines: Understanding genomic types of urinary bladder cancer. Tumour Biol. 2014, 35, 4599–4617. [Google Scholar] [CrossRef] [PubMed]
- Sturm, O.E.; Orton, R.; Grindlay, J.; Birtwistle, M.; Vyshemirsky, V.; Gilbert, D.; Calder, M.; Pitt, A.; Kholodenko, B.; Kolch, W. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci. Signal 2010, 3, ra90. [Google Scholar]
- Nawroth, R.; Stellwagen, F.; Schulz, W.A.; Stoehr, R.; Hartmann, A.; Krause, B.J.; Gschwend, J.E.; Retz, M. S6K1 and 4E-BP1 are independent regulated and control cellular growth in bladder cancer. PLoS One 2011, 6, e27509. [Google Scholar]
- Ramirez-Labrada, A.; Lopez-Royuela, N.; Jarauta, V.; Galan-Malo, P.; Azaceta, G.; Palomera, L.; Pardo, J.; Anel, A.; Marzo, I.; Naval, J. Two death pathways induced by sorafenib in myeloma cells: Puma-mediated apoptosis and necroptosis. Clin. Transl. Oncol. 2014. dio:10.1007/s12094-014-1201-y. Available online: http://link.springer.com/article/10.1007/s12094-014-1201-y (accessed on 19 July 2014).
- Llobet, D.; Eritja, N.; Yeramian, A.; Pallares, J.; Sorolla, A.; Domingo, M.; Santacana, M.; Gonzalez-Tallada, F.J.; Matias-Guiu, X.; Dolcet, X. The multikinase inhibitor sorafenib induces apoptosis and sensitises endometrial cancer cells to trail by different mechanisms. Eur. J. Cancer 2010, 46, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Bruzek, L.M.; Meng, X.W.; Gores, G.J.; Carter, C.A.; Kaufmann, S.H.; Adjei, A.A. The role of Mcl-11 downregulation in the proapoptotic activity of the multikinase inhibitor BAY 43-9006. Oncogene 2005, 24, 6861–6869. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Targeting c-FLICE-like inhibitory protein (CFLAR) in cancer. Expert Ther. Targets 2013, 17, 195–201. [Google Scholar] [CrossRef]
- Safa, A.R. C-FLIP, a master anti-apoptotic regulator. Exp. Oncol. 2012, 34, 176–184. [Google Scholar] [PubMed]
- Chan, S.L.; Yu, V.C. Proteins of the Bcl-2 family in apoptosis signalling: From mechanistic insights to therapeutic opportunities. Clin. Exp. Pharmacol. Physiol. 2004, 31, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Park, M.A.; Mitchell, C.; Hamed, H.; Rahmani, M.; Martin, A.P.; Curiel, D.T.; Yacoub, A.; Graf, M.; Lee, R.; et al. Vorinostat and sorafenib synergistically kill tumor cells via flip suppression and CD95 activation. Clin. Cancer Res. 2008, 14, 5385–5399. [Google Scholar] [CrossRef] [PubMed]
- Muhlethaler-Mottet, A.; Meier, R.; Flahaut, M.; Bourloud, K.B.; Nardou, K.; Joseph, J.M.; Gross, N. Complex molecular mechanisms cooperate to mediate histone deacetylase inhibitors anti-tumour activity in neuroblastoma cells. Mol. Cancer 2008, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Kretzner, L.; Scuto, A.; Dino, P.M.; Kowolik, C.M.; Wu, J.; Ventura, P.; Jove, R.; Forman, S.J.; Yen, Y.; Kirschbaum, M.H. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res. 2011, 71, 3912–3920. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, J.L.; Stasik, I.; Kerr, E.M.; Holohan, C.; Redmond, K.M.; McLaughlin, K.M.; Busacca, S.; Barbone, D.; Broaddus, V.C.; Gray, S.G.; et al. Vorinostat/SAHA-induced apoptosis in malignant mesothelioma is FLIP/caspase 8-dependent and HR23B-independent. Eur. J. Cancer 2012, 48, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Rosik, L.; Niegisch, G.; Fischer, U.; Jung, M.; Schulz, W.A.; Hoffmann, M.J. Limited efficacy of specific HDAC6 inhibition in urothelial cancer cells. Cancer Biol. Ther. 2014, 15, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Dumont, S.N.; Yang, D.; Dumont, A.G.; Reynoso, D.; Blay, J.Y.; Trent, J.C. Targeted polytherapy in small cell sarcoma and its association with doxorubicin. Mol. Oncol. 2014. [Google Scholar] [CrossRef]
- Park, M.A.; Zhang, G.; Martin, A.P.; Hamed, H.; Mitchell, C.; Hylemon, P.B.; Graf, M.; Rahmani, M.; Ryan, K.; Liu, X.; et al. Vorinostat and sorafenib increase er stress, autophagy and apoptosis via ceramide-dependent CD95 and perk activation. Cancer Biol. Ther. 2008, 7, 1648–1662. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, A.J.; Ma, S.L.; Cui, L.J.; Wu, B.; Yin, L.; Wu, M.C. Inhibition of autophagy significantly enhances combination therapy with sorafenib and HDAC inhibitors for human hepatoma cells. World J. Gastroenterol. 2014, 20, 4953–4962. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, D.E.; Jeong, I.G.; Choi, J.; Jang, S.; Lee, J.H.; Ro, S.; Hwang, J.J.; Kim, C.S. HDAC inhibitors synergize anti-proliferative effect of sorafenib in renal cell carcinoma cells. Anticancer Res. 2012, 32, 3161–3168. [Google Scholar] [PubMed]
- Perabo, F.G.; Kamp, S.; Schmidt, D.; Lindner, H.; Steiner, G.; Mattes, R.H.; Wirger, A.; Pegelow, K.; Albers, P.; Kohn, E.C.; et al. Bladder cancer cells acquire competent mechanisms to escape Fas-mediated apoptosis and immune surveillance in the course of malignant transformation. Br. J. Cancer 2001, 84, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.; Warskulat, U.; Steinhoff, C.; Mueller, W.; Grimm, M.O.; Schulz, W.A.; Seifert, H.H. Decreased Fas expression in advanced-stage bladder cancer is not related to p53 status. Urology 2004, 63, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Ewald, F.; Ueffing, N.; Brockmann, L.; Hader, C.; Telieps, T.; Schuster, M.; Schulz, W.A.; Schmitz, I. The role of c-FLIP splice variants in urothelial tumours. Cell Death Dis. 2011, 2, e245. [Google Scholar] [CrossRef]
- Hikita, H.; Takehara, T.; Shimizu, S.; Kodama, T.; Shigekawa, M.; Iwase, K.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Ishida, H.; et al. The Bcl-XL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology 2010, 52, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C., Jr.; Ferreira-Gonzalez, A.; Grant, S. Inhibition of Bcl-2 anti-apoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 2012, 119, 6089–6098. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes Mcl1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Seifert, H.H.; Meyer, A.; Cronauer, M.V.; Hatina, J.; Muller, M.; Rieder, H.; Hoffmann, M.J.; Ackermann, R.; Schulz, W.A. A new and reliable culture system for superficial low-grade urothelial carcinoma of the bladder. World J. Urol. 2007, 25, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.; Pohlmann, S.; Janicke, R.U.; Schulze-Osthoff, K.; Fischer, U. Apaf-1 and caspase-9 deficiency prevents apoptosis in a Bax-controlled pathway and promotes clonogenic survival during paclitaxel treatment. Blood 2007, 110, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; Liu, W.; Ju, Z.; Motter, T.; Peng, B.; Ryan, M.; et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knievel, J.; Schulz, W.A.; Greife, A.; Hader, C.; Lübke, T.; Schmitz, I.; Albers, P.; Niegisch, G. Multiple Mechanisms Mediate Resistance to Sorafenib in Urothelial Cancer. Int. J. Mol. Sci. 2014, 15, 20500-20517. https://doi.org/10.3390/ijms151120500
Knievel J, Schulz WA, Greife A, Hader C, Lübke T, Schmitz I, Albers P, Niegisch G. Multiple Mechanisms Mediate Resistance to Sorafenib in Urothelial Cancer. International Journal of Molecular Sciences. 2014; 15(11):20500-20517. https://doi.org/10.3390/ijms151120500
Chicago/Turabian StyleKnievel, Judith, Wolfgang A. Schulz, Annemarie Greife, Christiane Hader, Tobias Lübke, Ingo Schmitz, Peter Albers, and Günter Niegisch. 2014. "Multiple Mechanisms Mediate Resistance to Sorafenib in Urothelial Cancer" International Journal of Molecular Sciences 15, no. 11: 20500-20517. https://doi.org/10.3390/ijms151120500