Battle Against Cancer: An Everlasting Saga of p53



Abstract
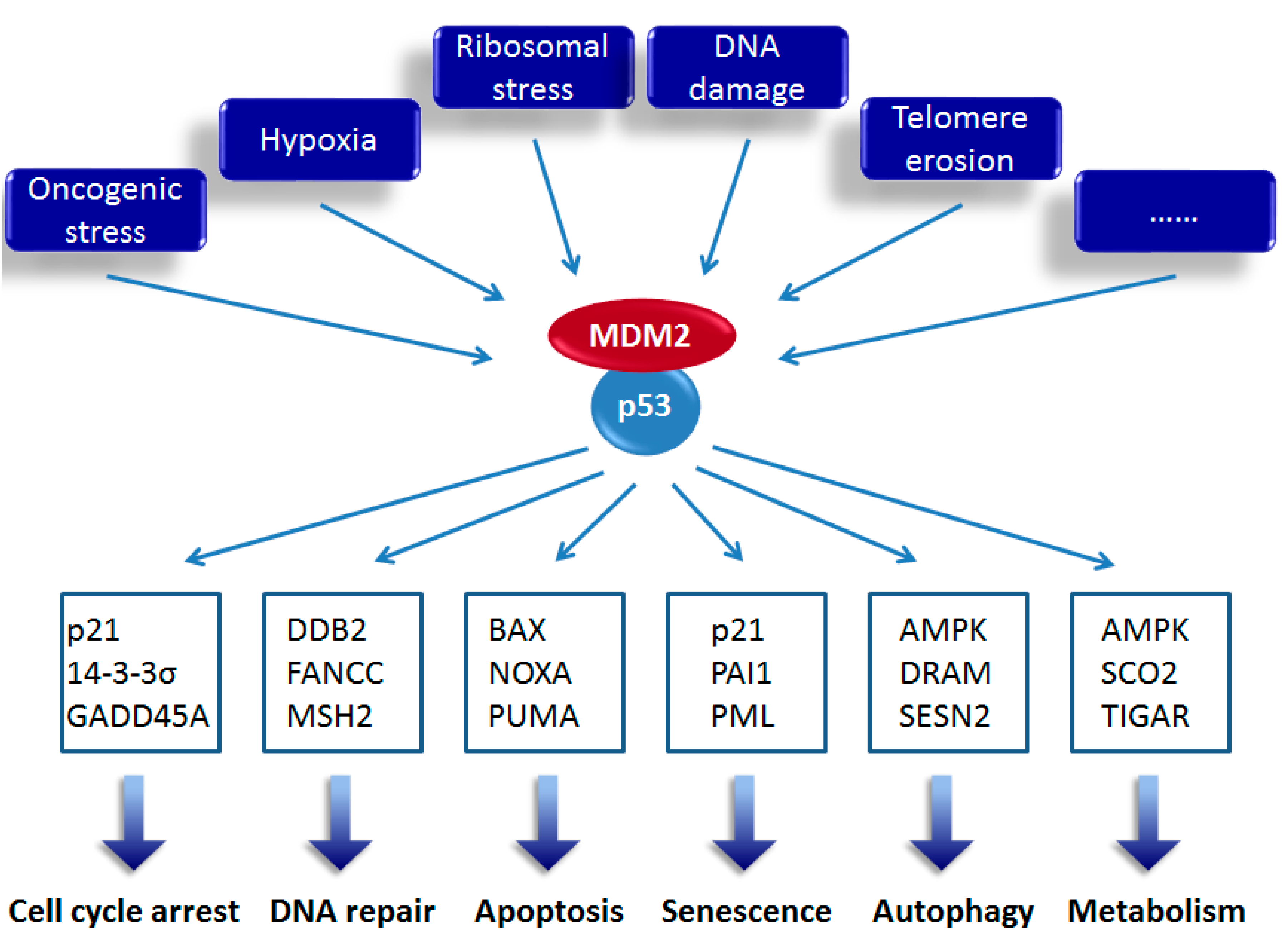
:1. Introduction
2. Activation of p53 upon Ribosomal Stress

3. Roles of p53 and Mutant p53 Target Genes
4. Drug Development

{kind=link}
| ID | Company | Status | Mechanism | In vivo Test | References |
|---|---|---|---|---|---|
| RG7112 (RO5045337) | Hoffmann-La Roche | Phase I | Inhibition of MDM2-p53 interaction | Advanced malignancies, except leukemia | [125] |
| MI-773 (SAR405838) | Sanofi-Aventis | Phase I | Inhibition of MDM2-p53 interaction | Advanced cancer | [126] |
| CGM097 | Novartis | Phase I | Inhibition of MDM2-p53 interaction | Selected advanced and refractory solid tumors | [126] |
| MK-8242 | Merck | Phase I | Inhibition of MDM2-p53 interaction | - | [126] |
| RO5503781 | Hoffmann-La Roche | Phase I | Inhibition of MDM2-p53 interaction | Soft tissue sarcoma; leukemia | [142] |
| Tenovin | - | Preclinical | Inhibition of Sirt1 and Sirt2 activity | - | [129] |
| Inauhzin | - | Preclinical | Inhibition of Sirt1 activity | - | [130] |
| PRIMA-1MET/APR-246 | Aprea AB | Phase I/II | Reactivate mutant p53 | Refractory hematologic malignancies and prostate cancer | [136] |
| PK083 | - | Preclinical | Reactivate mutant p53 Y220C | - | [138] |
| PK7088 | - | Preclinical | Reactivate mutant p53 Y220C | - | [139] |
| NSC319726 | - | Preclinical | Reactivate mutant p53 R175H | - | [140] |
5. Conclusions and Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar]
- Kress, M.; May, E.; Cassingena, R.; May, P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. Virology 1979, 31, 472–483. [Google Scholar]
- Melero, J.A.; Stitt, D.T.; Mangel, W.F.; Carroll, R.B. Identification of new polypeptide species (48–55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology 1979, 93, 466–480. [Google Scholar]
- Smith, A.E.; Smith, R.; Paucha, E. Characterization of different tumor antigens present in cells transformed by simian virus 40. Cell 1979, 18, 335–346. [Google Scholar]
- Eliyahu, D.; Raz, A.; Gruss, P.; Givol, D.; Oren, M. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 1984, 312, 646–649. [Google Scholar]
- Jenkins, J.R.; Rudge, K.; Currie, G.A. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature 1984, 312, 651–654. [Google Scholar]
- Parada, L.F.; Land, H.; Weinberg, R.A.; Wolf, D.; Rotter, V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 1984, 312, 649–651. [Google Scholar]
- Eliyahu, D.; Goldfinger, N.; Pinhasi-Kimhi, O.; Shaulsky, G.; Skurnik, Y.; Arai, N.; Rotter, V.; Oren, M. Meth A fibrosarcoma cells express two transforming mutant p53 species. Oncogene 1988, 3, 313–321. [Google Scholar]
- Finlay, C.A.; Hinds, P.W.; Tan, T.H.; Eliyahu, D.; Oren, M.; Levine, A.J. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol. Cell. Biol. 1988, 8, 531–539. [Google Scholar]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; van Tuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar]
- Marchenko, N.D.; Moll, U.M. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle 2007, 6, 1718–1723. [Google Scholar]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar]
- Poyurovsky, M.V.; Katz, C.; Laptenko, O.; Beckerman, R.; Lokshin, M.; Ahn, J.; Byeon, I.J.; Gabizon, R.; Mattia, M.; Zupnick, A; et al. The C terminus of p53 binds the N-terminal domain of MDM2. Nat. Struct. Mol. Biol. 2010, 17, 982–989. [Google Scholar]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar]
- Fuchs, S.Y.; Adler, V.; Buschmann, T.; Wu, X.; Ronai, Z. Mdm2 association with p53 targets its ubiquitination. Oncogene 1998, 17, 2543–2547. [Google Scholar]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar]
- Zhao, H.; Bauzon, F.; Fu, H.; Lu, Z.; Cui, J.; Nakayama, K.; Nakayama, K.I.; Locker, J.; Zhu, L. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell 2013, 24, 645–659. [Google Scholar]
- Appella, E.; Anderson, C.W. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 2001, 268, 2764–2772. [Google Scholar]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar]
- Liu, L.; Scolnick, D.M.; Trievel, R.C.; Zhang, H.B.; Marmorstein, R.; Halazonetis, T.D.; Berger, S.L. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 1999, 19, 1202–1209. [Google Scholar]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar]
- An, W.G.; Kanekal, M.; Simon, M.C.; Maltepe, E.; Blagosklonny, M.V.; Neckers, L.M. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature 1998, 392, 405–408. [Google Scholar]
- Hammond, E.M.; Denko, N.C.; Dorie, M.J.; Abraham, R.T.; Giaccia, A.J. Hypoxia links ATR and p53 through replication arrest. Mol. Cell. Biol. 2002, 22, 1834–1843. [Google Scholar]
- Lee, J.H.; Jin, Y.; He, G.; Zeng, S.X.; Wang, Y.V.; Wahl, G.M.; Lu, H. Hypoxia activates tumor suppressor p53 by inducing ATR-Chk1 kinase cascade-mediated phosphorylation and consequent 14-3-3 gamma inactivation of MDMX protein. J. Biol. Chem. 2012, 287, 20898–20903. [Google Scholar]
- Zhou, X.; Liao, J.M.; Liao, W.J.; Lu, H. Scission of the p53-MDM2 Loop by Ribosomal Proteins. Genes Cancer 2012, 3, 298–310. [Google Scholar]
- Marechal, V.; Elenbaas, B.; Piette, J.; Nicolas, J.C.; Levine, A.J. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol. Cell. Biol. 1994, 14, 7414–7420. [Google Scholar]
- Lohrum, M.A.; Ludwig, R.L.; Kubbutat, M.H.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003, 23, 8902–8912. [Google Scholar]
- Dai, M.S.; Lu, H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar]
- Dai, M.S.; Zeng, S.X.; Jin, Y.; Sun, X.X.; David, L.; Lu, H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell. Biol. 2004, 24, 7654–7668. [Google Scholar]
- Jin, A.; Itahana, K.; O’Keefe, K.; Zhang, Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell. Biol. 2004, 24, 7669–7680. [Google Scholar]
- Bai, D.; Zhang, J.; Xiao, W.; Zheng, X. Regulation of the HDM2-p53 pathway by ribosomal protein L6 in response to ribosomal stress. Nucleic Acids Res. 2014, 42, 1799–1811. [Google Scholar]
- Zhang, Y.; Wang, J.; Yuan, Y.; Zhang, W.; Guan, W.; Wu, Z.; Jin, C.; Chen, H.; Zhang, L.; Yang, X.; et al. Negative regulation of HDM2 to attenuate p53 degradation by ribosomal protein L26. Nucleic Acids Res. 2010, 38, 6544–6554. [Google Scholar]
- Daftuar, L.; Zhu, Y.; Jacq, X.; Prives, C. Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2-p53-MdmX network. PLoS One 2013, 8, e68667. [Google Scholar]
- Yadavilli, S.; Mayo, L.D.; Higgins, M.; Lain, S.; Hegde, V.; Deutsch, W.A. Ribosomal protein S3: A multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair 2009, 8, 1215–1224. [Google Scholar]
- Zhu, Y.; Poyurovsky, M.V.; Li, Y.; Biderman, L.; Stahl, J.; Jacq, X.; Prives, C. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol. Cell 2009, 35, 316–326. [Google Scholar]
- Chen, D.; Zhang, Z.; Li, M.; Wang, W.; Li, Y.; Rayburn, E.R.; Hill, D.L.; Wang, H.; Zhang, R. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: Binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene 2007, 26, 5029–5037. [Google Scholar]
- Zhou, X.; Hao, Q.; Liao, J.; Zhang, Q.; Lu, H. Ribosomal protein S14 unties the MDM2-p53 loop upon ribosomal stress. Oncogene 2013, 32, 388–396. [Google Scholar]
- Zhang, X.; Wang, W.; Wang, H.; Wang, M.H.; Xu, W.; Zhang, R. Identification of ribosomal protein S25 (RPS25)-MDM2-p53 regulatory feedback loop. Oncogene 2013, 32, 2782–2791. [Google Scholar]
- Cui, D.; Li, L.; Lou, H.; Sun, H.; Ngai, S.M.; Shao, G.; Tang, J. The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 2014, 33, 2225–2235. [Google Scholar]
- Xiong, X.; Zhao, Y.; He, H.; Sun, Y. Ribosomal protein S27-like and S27 interplay with p53-MDM2 axis as a target, a substrate and a regulator. Oncogene 2011, 30, 1798–1811. [Google Scholar]
- Sun, X.X.; DeVine, T.; Challagundla, K.B.; Dai, M.S. Interplay between ribosomal protein S27a and MDM2 protein in p53 activation in response to ribosomal stress. J. Biol. Chem. 2011, 286, 22730–22741. [Google Scholar]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar]
- Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; Kastan, M.B.; Oren, M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 2008, 32, 180–189. [Google Scholar]
- Mahata, B.; Sundqvist, A.; Xirodimas, D.P. Recruitment of RPL11 at promoter sites of p53-regulated genes upon nucleolar stress through NEDD8 and in an Mdm2-dependent manner. Oncogene 2012, 31, 3060–3071. [Google Scholar]
- Macias, E.; Jin, A.; Deisenroth, C.; Bhat, K.; Mao, H.; Lindstrom, M.S.; Zhang, Y. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 2010, 18, 231–243. [Google Scholar]
- Lindstrom, M.S.; Jin, A.; Deisenroth, C.; White Wolf, G.; Zhang, Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol. Cell. Biol. 2007, 27, 1056–1068. [Google Scholar]
- Sasaki, M.; Kawahara, K.; Nishio, M.; Mimori, K.; Kogo, R.; Hamada, K.; Itoh, B.; Wang, J.; Komatsu, Y.; Yang, Y.R.; et al. Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 2011, 17, 944–951. [Google Scholar]
- Fregoso, O.I.; Das, S.; Akerman, M.; Krainer, A.R. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol. Cell 2013, 50, 56–66. [Google Scholar]
- Dai, M.S.; Challagundla, K.B.; Sun, X.X.; Palam, L.R.; Zeng, S.X.; Wek, R.C.; Lu, H. Physical and functional interaction between ribosomal protein L11 and the tumor suppressor ARF. J. Biol. Chem. 2012, 287, 17120–17129. [Google Scholar]
- Kino, K.; Yamashita, A.; Yamaoka, K.; Watanabe, J.; Tanaka, S.; Ko, K.; Shimizu, K.; Tsunoo, H. Isolation and characterization of a new immunomodulatory protein, ling zhi-8 (LZ-8), from Ganoderma lucidium. J. Biol. Chem. 1989, 264, 472–478. [Google Scholar]
- Wu, C.T.; Lin, T.Y.; Hsu, H.Y.; Sheu, F.; Ho, C.M.; Chen, E.I. Ling Zhi-8 mediates p53-dependent growth arrest of lung cancer cells proliferation via the ribosomal protein S7-MDM2-p53 pathway. Carcinogenesis 2011, 32, 1890–1896. [Google Scholar]
- Sundqvist, A.; Liu, G.; Mirsaliotis, A.; Xirodimas, D.P. Regulation of nucleolar signalling to p53 through NEDDylation of L11. EMBO Rep. 2009, 10, 1132–1139. [Google Scholar]
- Zhang, J.; Bai, D.; Ma, X.; Guan, J.; Zheng, X. hCINAP is a novel regulator of ribosomal protein-HDM2-p53 pathway by controlling NEDDylation of ribosomal protein S14. Oncogene 2014, 33, 246–254. [Google Scholar]
- Wilson-Edell, K.A.; Kehasse, A.; Scott, G.K.; Yau, C.; Rothschild, D.E.; Schilling, B.; Gabriel, B.S.; Yevtushenko, M.A.; Hanson, I.M.; Held, J.M.; et al. RPL24: A potential therapeutic target whose depletion or acetylation inhibits polysome assembly and cancer cell growth. Oncotarget 2014, 5, 5165–5176. [Google Scholar]
- Ventimiglia, F.A.; Wool, I.G. A kinase that transfers the gamma-phosphoryl group of GTP to proteins of eukaryotic 40S ribosomal subunits. Proc. Nat. Acad. Sci. USA 1974, 71, 350–354. [Google Scholar]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar]
- Funk, W.D.; Pak, D.T.; Karas, R.H.; Wright, W.E.; Shay, J.W. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol. 1992, 12, 2866–2871. [Google Scholar]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar]
- Carvajal, L.A.; Manfredi, J.J. Another fork in the road—life or death decisions by the tumour suppressor p53. EMBO Rep. 2013, 14, 414–421. [Google Scholar]
- Chen, X.; Ko, L.J.; Jayaraman, L.; Prives, C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 1996, 10, 2438–2451. [Google Scholar]
- Resnick-Silverman, L.; St Clair, S.; Maurer, M.; Zhao, K.; Manfredi, J.J. Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev. 1998, 12, 2102–2107. [Google Scholar]
- Inga, A.; Storici, F.; Darden, T.A.; Resnick, M.A. Differential transactivation by the p53 transcription factor is highly dependent on p53 level and promoter target sequence. Mol. Cell. Biol. 2002, 22, 8612–8625. [Google Scholar]
- Weinberg, R.L.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005, 348, 589–596. [Google Scholar]
- Oda, K.; Arakawa, H.; Tanaka, T.; Matsuda, K.; Tanikawa, C.; Mori, T.; Nishimori, H.; Tamai, K.; Tokino, T.; Nakamura, Y.; et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 2000, 102, 849–862. [Google Scholar]
- Samuels-Lev, Y.; O’Connor, D.J.; Bergamaschi, D.; Trigiante, G.; Hsieh, J.K.; Zhong, S.; Campargue, I.; Naumovski, L.; Crook, T.; Lu, X. ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 2001, 8, 781–794. [Google Scholar]
- Bergamaschi, D.; Samuels, Y.; Sullivan, A.; Zvelebil, M.; Breyssens, H.; Bisso, A.; del Sal, G.; Syed, N.; Smith, P.; Gasco, M.; et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat. Genet. 2006, 38, 1133–1141. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- He, L.; He, X.; Lowe, S.W.; Hannon, G.J. microRNAs join the p53 network-another piece in the tumour-suppression puzzle. Nat. Rev. Cancer 2007, 7, 819–822. [Google Scholar]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar]
- Liao, J.M.; Cao, B.; Zhou, X.; Lu, H. New insights into p53 functions through its target microRNAs. J. Mol. Cell Biol. 2014, 6, 206–213. [Google Scholar]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar]
- Christoffersen, N.R.; Shalgi, R.; Frankel, L.B.; Leucci, E.; Lees, M.; Klausen, M.; Pilpel, Y.; Nielsen, F.C.; Oren, M.; Lund, A.H. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010, 17, 236–245. [Google Scholar]
- Tazawa, H.; Tsuchiya, N.; Izumiya, M.; Nakagama, H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Nat. Acad. Sci. USA 2007, 104, 15472–15477. [Google Scholar]
- Kim, N.H.; Kim, H.S.; Kim, N.G.; Lee, I.; Choi, H.S.; Li, X.Y.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Na, J.M.; et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal. 2011, 4. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar]
- Weeraratne, S.D.; Amani, V.; Neiss, A.; Teider, N.; Scott, D.K.; Pomeroy, S.L.; Cho, Y.J. miR-34a confers chemosensitivity through modulation of MAGE-A and p53 in medulloblastoma. Neuro-Oncology 2011, 13, 165–175. [Google Scholar]
- Siemens, H.; Jackstadt, R.; Kaller, M.; Hermeking, H. Repression of c-Kit by p53 is mediated by miR-34 and is associated with reduced chemoresistance, migration and stemness. Oncotarget 2013, 4, 1399–1415. [Google Scholar]
- Zhang, A.; Xu, M.; Mo, Y.Y. Role of the lncRNA-p53 regulatory network in cancer. J. Mol. Cell Biol. 2014, 6, 181–191. [Google Scholar]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar]
- Chan, W.M.; Siu, W.Y.; Lau, A.; Poon, R.Y. How many mutant p53 molecules are needed to inactivate a tetramer? Mol. Cell. Biol. 2004, 24, 3536–3551. [Google Scholar]
- Xu, J.; Reumers, J.; Couceiro, J.R.; de Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar]
- Marin, M.C.; Jost, C.A.; Brooks, L.A.; Irwin, M.S.; O’Nions, J.; Tidy, J.A.; James, N.; McGregor, J.M.; Harwood, C.A.; Yulug, I.G.; et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat. Genet. 2000, 25, 47–54. [Google Scholar]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826. [Google Scholar]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar]
- Bargonetti, J.; Chicas, A.; White, D.; Prives, C. p53 represses Sp1 DNA binding and HIV-LTR directed transcription. Cell. Mol. Biol. 1997, 43, 935–949. [Google Scholar]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar]
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012, 26, 830–845. [Google Scholar]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar]
- Gohler, T.; Jager, S.; Warnecke, G.; Yasuda, H.; Kim, E.; Deppert, W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res. 2005, 33, 1087–1100. [Google Scholar]
- Brazdova, M.; Quante, T.; Togel, L.; Walter, K.; Loscher, C.; Tichy, V.; Cincarova, L.; Deppert, W.; Tolstonog, G.V. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res. 2009, 37, 1486–1500. [Google Scholar]
- Liu, K.; Ling, S.; Lin, W.C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell. Biol. 2011, 31, 4464–4481. [Google Scholar]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar]
- Zak, K.; Pecak, A.; Rys, B.; Wladyka, B.; Domling, A.; Weber, L.; Holak, T.A.; Dubin, G. Mdm2 and MdmX inhibitors for the treatment of cancer: A patent review (2011–present). Expert Opin. Ther. Pat. 2013, 23, 425–448. [Google Scholar]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [Google Scholar]
- Zhang, Q.; Zeng, S.X.; Zhang, Y.; Ding, D.; Ye, Q.; Meroueh, S.O.; Lu, H. A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol. Med. 2012, 4, 298–312. [Google Scholar]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maiga, S.; Lode, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar]
- Peng, X.; Zhang, M.Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.; Arner, E.S.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Rokaeus, N.; Shen, J.; Eckhardt, I.; Bykov, V.J.; Wiman, K.G.; Wilhelm, M.T. PRIMA-1(MET)/APR-246 targets mutant forms of p53 famly members p63 and p73. Oncogene 2010, 29, 6442–6451. [Google Scholar]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andren, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Nat. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar]
- Zhang, Q.; Zeng, S.X.; Lu, H. Targeting p53-MDM2-MDMX Loop for Cancer Therapy. Subcell. Biochem. 2014, 85, 281–319. [Google Scholar]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Kenzelmann Broz, D.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar]
- Campisi, J. Parsing p53 Transactivation. Dev. Cell 2011, 20, 573–574. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Q.; Cho, W.C. Battle Against Cancer: An Everlasting Saga of p53. Int. J. Mol. Sci. 2014, 15, 22109-22127. https://doi.org/10.3390/ijms151222109
Hao Q, Cho WC. Battle Against Cancer: An Everlasting Saga of p53. International Journal of Molecular Sciences. 2014; 15(12):22109-22127. https://doi.org/10.3390/ijms151222109
Chicago/Turabian StyleHao, Qian, and William C. Cho. 2014. "Battle Against Cancer: An Everlasting Saga of p53" International Journal of Molecular Sciences 15, no. 12: 22109-22127. https://doi.org/10.3390/ijms151222109