The Emerging Nexus of Active DNA Demethylation and Mitochondrial Oxidative Metabolism in Post-Mitotic Neurons

Abstract
:
1. Introduction
2. DNA Methylation Is Stable and Reversible

3. TET (Ten-Eleven Translocation) Dioxygenases Link Metabolic Intermediates to Active DNA Demethylation
4. ROS (Reactive Oxygen Species) and Oxidizing Agents Modify Methylated Cytosine Derivatives

5. Active DNA Demethylation in Post-Mitotic Neurons
6. TET Methylcytosine Dioxygenases in Memory Formation and Reversal Learning

{kind=link}
{kind=link}
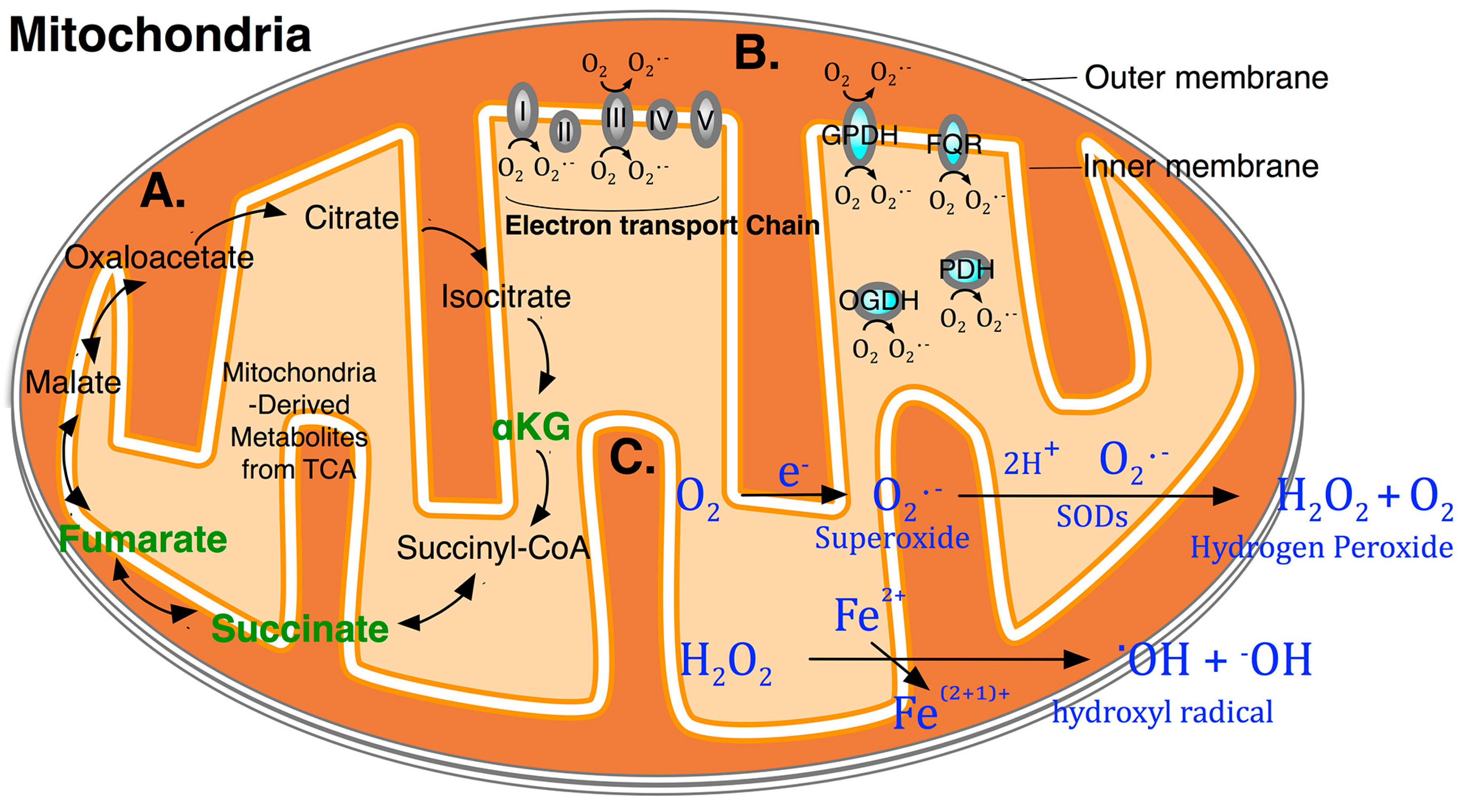{kind=link}
{kind=link}
| Tet Dioxygenases | Transcription in Adult Mouse Brain [61] | Known DNA Substrates | Major Mouse Phenotypes Partially from Mouse Genome Informatics (MGI) |
|---|---|---|---|
| Tet1 | Cerebellum: medium Cortex: low Hippocampus: low | 5mC to 5hmC T to 5hmU [32] 5hmC to 5fC 5fC to 5caC | Knockout mice are viable, fertile and grossly normal. Some mutant mice have mild embryonic growth retardation, decreased body size and small litters [ 62]. One line of knockout mice exhibits abnormal hippocampal long-term depression and impaired memory extinction [58]. Another line of knockout mice exhibits impaired hippocampal neurogenesis accompanied by poor learning and memory [59]. |
| Tet2 | Cerebellum: medium Cortex: medium Hippocampus: medium | 5mC to 5hmC 5hmC to 5fC 5fC to 5caC | Knockout mice evolved to a wide spectrum of lethal myeloid malignancies [63]. Conditional knockout of hematopoietic compartment exhibits increased stem cell self-renewal and myeloid transformation [64–66]. |
| Tet3 | Cerebellum: high Cortex: high Hippocampus: high | 5mC to 5hmC 5hmC to 5fC 5fC to 5caC | Conditional knockout mice show impaired reprogramming of the paternal genome, resulting in reduced embryo viability. Female germ-line knockout mice show severely reduced fecundity and some of their heterozygous mutant offspring have increased developmental failure [67]. Cortical knockdown in mice shows impairment in fear extinction memory [57]. |

7. Implications for Potential Therapies of Neurodegeneration
| Agent Name | Mechanism | Clinical Trials [73] | Concerns and Notes [79] |
|---|---|---|---|
| 5-azacytidine | Cytosine analogue, suicide inhibitor of Dnmt | Clinically Tested: YES | Hematological malignancies, dose-limiting toxicity; Covalent DNA-protein trapping |
| FDA-approved: YES | |||
| Crosses BBB: NO | |||
| 5-aza-2'-deoxycytidine (Decitabine, 5-azadC) | Cytosine analogue, suicide inhibitor of Dnmt | Clinically Tested: YES | Same as above |
| FDA-approved: YES | |||
| Crosses BBB: NO | |||
| Procainamide (Pronestyl) | Acts on Dnmt to reduce its affinity, non-nucleoside, blocks sodium channels and a specific inhibitor of Dnmt1 [77] | Clinically Tested: YES | Cardiac arrhythmia, sodium channel blocker; drug-induced lupus erythematosus |
| FDA-approved: YES | |||
| Crosses BBB: YES | |||
| (–)-epigallocatechin-3-O-gallate (EGCG) | Direct inhibition of Dnmt by reducing its affinity, non-nucleoside | Clinically Tested: YES | Strong topoisomerase inhibitor; should not be used by pregnant women because of increased risk of neonatal leukaemia and childhood malignant CNS tumours |
| FDA-approved: NO | |||
| Crosses BBB: YES | |||
| RG108 | Direct inhibition of Dnmt by reducing its affinity, non-nucleoside and a specific inhibitor of Dnmt1 [78] | Clinically Tested: NO | Low concentration results in significant demethylation of genomic DNA without any detectable toxicity; preclinical for cancer chemotherapy and ALS therapy |
| FDA-approved: NO | |||
| Crosses BBB: UNKNOWN | |||
| Zebularine | Cytosine analogue | Clinically Tested: NO | Can be used orally; Lower toxicity than 5-azaC |
| FDA-approved: NO | |||
| Crosses BBB: NO | |||
| Hydralazine | Cytosine analogue | Clinically Tested: YES | Sympathetic stimulation of the heart; used successfully for myelodysplastic syndrome as a DNA methylation inhibitor |
| FDA-approved: YES | |||
| Crosses BBB: NO |
Acknowledgments
Conflicts of Interest
References
- Sproul, D.; Meehan, R.R. Genomic insights into cancer-associated aberrant CpG island hypermethylation. Brief Funct. Genomics 2013, 12, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.A.; Reddington, J.P.; Nestor, C.E.; Dunican, D.S.; Branco, M.R.; Reichmann, J.; Reik, W.; Surani, M.A.; Adams, I.R.; Meehan, R.R. Promoter DNA methylation couples genome-defence mechanisms to epigenetic reprogramming in the mouse germline. Development 2012, 139, 3623–3632. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner TET1. Science 2009, 324, 930–935. [Google Scholar]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Shen, L.; Dai, Q.; He, C.; Zhang, Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011, 21, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Song, C.X.; Szulwach, K.E.; Dai, Q.; Fu, Y.; Mao, S.Q.; Lin, L.; Street, C.; Li, Y.; Poidevin, M.; Wu, H.; et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 2013, 153, 678–691. [Google Scholar]
- Hashimoto, H.; Zhang, X.; Cheng, X. Excision of thymine and 5-hydroxymethyluracil by the mbd4 DNA glycosylase domain: Structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012, 40, 8276–8284. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar]
- Reddington, J.P.; Pennings, S.; Meehan, R.R. Non-canonical functions of the DNA methylome in gene regulation. Biochem. J. 2013, 451, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Reddington, J.P.; Perricone, S.M.; Nestor, C.E.; Reichmann, J.; Youngson, N.A.; Suzuki, M.; Reinhardt, D.; Dunican, D.S.; Prendegast, J.G.; Mjoseng, H.; et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb-target genes. Genome Biol. 2013, 14. [Google Scholar] [CrossRef]
- Jeltsch, A. Oxygen, epigenetic signaling, and the evolution of early life. Trends Biochem. Sci. 2013, 38, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Song, C.X.; He, C.; Zhang, Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu. Rev. Biochem. 2014, 83, 585–614. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Kangaspeska, S.; Stride, B.; Metivier, R.; Polycarpou-Schwarz, M.; Ibberson, D.; Carmouche, R.P.; Benes, V.; Gannon, F.; Reid, G. Transient cyclical methylation of promoter DNA. Nature 2008, 452, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Metivier, R.; Gallais, R.; Tiffoche, C.; le Peron, C.; Jurkowska, R.Z.; Carmouche, R.P.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G.; et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 452, 45–50. [Google Scholar]
- Thillainadesan, G.; Chitilian, J.M.; Isovic, M.; Ablack, J.N.; Mymryk, J.S.; Tini, M.; Torchia, J. TGF-β-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol. Cell 2012, 46, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar]
- Jacobs, A.L.; Schar, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.; Huggins, I.J.; James, S.R.; Karpf, A.R.; Jones, D.A.; Cairns, B.R. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 2008, 135, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Bransteitter, R.; Pham, P.; Scharff, M.D.; Goodman, M.F. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of rnase. Proc. Natl. Acad. Sci. USA 2003, 100, 4102–4107. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by tdg in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar]
- Nabel, C.S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H.L.; Stivers, J.T.; Zhang, Y.; Kohli, R.M. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 2012, 8, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Pfaffeneder, T.; Spada, F.; Wagner, M.; Brandmayr, C.; Laube, S.K.; Eisen, D.; Truss, M.; Steinbacher, J.; Hackner, B.; Kotljarova, O.; et al. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat. Chem. Biol. 2014, 10, 574–581. [Google Scholar]
- Krebs, C.; Galonic Fujimori, D.; Walsh, C.T.; Bollinger, J.M., Jr. Non-heme Fe(IV)-oxo intermediates. Acc. Chem. Res. 2007, 40, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, II; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. d-2-hydroxyglutarate produced by mutant idh1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Iqbal, K.; Jin, S.G.; Pfeifer, G.P.; Szabo, P.E. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. USA 2011, 108, 3642–3647. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.P.; Gavin, D.P.; Grayson, D.R. CpG methylation in neurons: Message, memory, or mask? Neuropsychopharmacology 2010, 35, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Jang, M.H.; Guo, J.U.; Kitabatake, Y.; Chang, M.L.; Pow-Anpongkul, N.; Flavell, R.A.; Lu, B.; Ming, G.L.; Song, H. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 2009, 323, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Thomassin, H.; Flavin, M.; Espinas, M.L.; Grange, T. Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J. 2001, 20, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Cervoni, N.; Szyf, M. Demethylase activity is directed by histone acetylation. J. Biol. Chem. 2001, 276, 40778–40787. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Meisel, A.; Biniszkiewicz, D.; Namura, S.; Prass, K.; Ruscher, K.; Lipski, A.; Jaenisch, R.; Moskowitz, M.A.; Dirnagl, U. DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 2000, 20, 3175–3181. [Google Scholar] [PubMed]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Sweatt, J.D. DNA methylation and memory formation. Nat. Neurosci. 2010, 13, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Campbell, S.L.; Sweatt, J.D. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol. Learn. Mem. 2008, 89, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Baker-Andresen, D.; Ratnu, V.S.; Bredy, T.W. Dynamic DNA methylation: A prime candidate for genomic metaplasticity and behavioral adaptation. Trends Neurosci. 2013, 36, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, W.; Ratnu, V.S.; Bredy, T.W. On the potential role of active DNA demethylation in establishing epigenetic states associated with neural plasticity and memory. Neurobiol. Learn. Mem. 2013, 105, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.L.; King, J.R.; Song, H.; Sweatt, J.D. Tet1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, W.; Zhao, Q.Y.; Widagdo, J.; Baker-Andresen, D.; Flavell, C.R.; D’Alessio, A.; Zhang, Y.; Bredy, T.W. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc. Natl. Acad. Sci. USA 2014, 111, 7120–7125. [Google Scholar] [CrossRef]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.H. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Cui, Q.Y.; Murai, K.; Lim, Y.C.; Smith, Z.D.; Jin, S.; Ye, P.; Rosa, L.; Lee, Y.K.; Wu, H.P.; et al. Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell 2013, 13, 237–245. [Google Scholar]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-methylcytosine by Tet1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Szwagierczak, A.; Bultmann, S.; Schmidt, C.S.; Spada, F.; Leonhardt, H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010, 38. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Ganz, K.; Powell, B.E.; Hu, Y.C.; Markoulaki, S.; Cheng, A.W.; Gao, Q.; Kim, J.; Choi, S.W.; Page, D.C.; et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 2011, 9, 166–175. [Google Scholar]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Quivoron, C.; Couronne, L.; della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. Tet2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar]
- Gu, T.P.; Guo, F.; Yang, H.; Wu, H.P.; Xu, G.F.; Liu, W.; Xie, Z.G.; Shi, L.; He, X.; Jin, S.G.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar]
- Day, J.J.; Sweatt, J.D. Epigenetic mechanisms in cognition. Neuron 2011, 70, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Champagne, F.A.; Curley, J.P. Epigenetic mechanisms mediating the long-term effects of maternal care on development. Neurosci. Biobehav. Rev. 2009, 33, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Dulac, C. Brain function and chromatin plasticity. Nature 2010, 465, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Nestler, E.J. Epigenetic mechanisms in drug addiction. Trends Mol. Med. 2008, 14, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Gavin, D.P.; Chase, K.A.; Sharma, R.P. Active DNA demethylation in post-mitotic neurons: A reason for optimism. Neuropharmacology 2013, 75, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.P.; Chase, K.A. Increasing neuronal “stemness”: Chromatin relaxation and the expression of reprogramming genes in post-mitotic neurons. Med. Hypotheses 2012, 78, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Sigalotti, L.; Covre, A.; Fratta, E.; Parisi, G.; Colizzi, F.; Rizzo, A.; Danielli, R.; Nicolay, H.J.; Coral, S.; Maio, M. Epigenetics of human cutaneous melanoma: Setting the stage for new therapeutic strategies. J. Transl. Med. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Yegnasubramanian, S.; Lin, X.; Nelson, W.G. Procainamide is a specific inhibitor of DNA methyltransferase 1. J. Biol. Chem. 2005, 280, 40749–40756. [Google Scholar] [CrossRef] [PubMed]
- Asgatay, S.; Champion, C.; Marloie, G.; Drujon, T.; Senamaud-Beaufort, C.; Ceccaldi, A.; Erdmann, A.; Rajavelu, A.; Schambel, P.; Jeltsch, A.; et al. Synthesis and evaluation of analogues of N-phthaloyl-l-tryptophan (RG108) as inhibitors of DNA methyltransferase 1. J. Med. Chem. 2014, 57, 421–434. [Google Scholar]
- Martin, L.J.; Wong, M. Aberrant regulation of DNA methylation in amyotrophic lateral sclerosis: A new target of disease mechanisms. Neurotherapeutics 2013, 10, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Klisovic, R.B.; Stock, W.; Cataland, S.; Klisovic, M.I.; Liu, S.; Blum, W.; Green, M.; Odenike, O.; Godley, L.; Burgt, J.V.; et al. A phase I biological study of MG98, an oligodeoxynucleotide antisense to DNA methyltransferase 1, in patients with high-risk myelodysplasia and acute myeloid leukemia. Clin. Cancer Res. 2008, 14, 2444–2449. [Google Scholar]
- Winquist, E.; Knox, J.; Ayoub, J.P.; Wood, L.; Wainman, N.; Reid, G.K.; Pearce, L.; Shah, A.; Eisenhauer, E. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: A national cancer institute of canada clinical trials group investigational new drug study. Investig. New Drugs 2006, 24, 159–167. [Google Scholar] [CrossRef]
- Davis, A.J.; Gelmon, K.A.; Siu, L.L.; Moore, M.J.; Britten, C.D.; Mistry, N.; Klamut, H.; D’Aloisio, S.; MacLean, M.; Wainman, N.; et al. Phase I and pharmacologic study of the human DNA methyltransferase antisense oligodeoxynucleotide MG98 given as a 21-day continuous infusion every 4 weeks. Investig. New Drugs 2003, 21, 85–97. [Google Scholar]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Felle, M.; Joppien, S.; Nemeth, A.; Diermeier, S.; Thalhammer, V.; Dobner, T.; Kremmer, E.; Kappler, R.; Langst, G. The USP7/Dnmt1 complex stimulates the DNA methylation activity of dnmt1 and regulates the stability of UHRF1. Nucleic Acids Res. 2011, 39, 8355–8365. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Chen, H.; Guo, X.; Wang, Z.; Sowa, M.E.; Zheng, L.; Hu, S.; Zeng, P.; Guo, R.; Diao, J.; et al. M phase phosphorylation of the epigenetic regulator UHRF1 regulates its physical association with the deubiquitylase USP7 and stability. Proc. Natl. Acad. Sci. USA 2012, 109, 4828–4833. [Google Scholar]
- Qin, W.; Leonhardt, H.; Spada, F. USP7 and UHRF1 control ubiquitination and stability of the maintenance DNA methyltransferase DNMT1. J. Cell. Biochem. 2011, 112, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar]
- Meng, H.; Harrison, D.J.; Meehan, R.R. MBD4 interacts with and recruits USP7 to heterochromatic foci. J. Cell. Biochem. 2014. [Google Scholar] [CrossRef]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Aijo, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lahdesmaki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013, 497, 122–126. [Google Scholar]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Abhiman, S.; Aravind, L. Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 2011, 101, 25–104. [Google Scholar] [PubMed]
- Xu, Y.; Wu, F.; Tan, L.; Kong, L.; Xiong, L.; Deng, J.; Barbera, A.J.; Zheng, L.; Zhang, H.; Huang, S.; et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by TET1 hydroxylase in mouse embryonic stem cells. Mol. Cell 2011, 42, 451–464. [Google Scholar]
- Jin, C.; Lu, Y.; Jelinek, J.; Liang, S.; Estecio, M.R.; Barton, M.C.; Issa, J.P. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014, 42, 6956–6971. [Google Scholar] [CrossRef] [PubMed]
- Vella, P.; Scelfo, A.; Jammula, S.; Chiacchiera, F.; Williams, K.; Cuomo, A.; Roberto, A.; Christensen, J.; Bonaldi, T.; Helin, K.; et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell 2013, 49, 645–656. [Google Scholar]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Ait-Si-Ali, S.; Yokochi, T.; Wade, P.A.; Jones, P.L.; Wolffe, A.P. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 2000, 25, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.; Critchlow, S.E. Emerging approaches to target tumor metabolism. Curr. Opin. Pharmacol. 2014, 17C, 22–29. [Google Scholar] [CrossRef]
- Gut, P.; Verdin, E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013, 502, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell MeTab 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 2013, 31, 1137–1142. [Google Scholar]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, H.; Chen, G.; Gao, H.-M.; Song, X.; Shi, Y.; Cao, L. The Emerging Nexus of Active DNA Demethylation and Mitochondrial Oxidative Metabolism in Post-Mitotic Neurons. Int. J. Mol. Sci. 2014, 15, 22604-22625. https://doi.org/10.3390/ijms151222604
Meng H, Chen G, Gao H-M, Song X, Shi Y, Cao L. The Emerging Nexus of Active DNA Demethylation and Mitochondrial Oxidative Metabolism in Post-Mitotic Neurons. International Journal of Molecular Sciences. 2014; 15(12):22604-22625. https://doi.org/10.3390/ijms151222604
Chicago/Turabian StyleMeng, Huan, Guiquan Chen, Hui-Ming Gao, Xiaoyu Song, Yun Shi, and Liu Cao. 2014. "The Emerging Nexus of Active DNA Demethylation and Mitochondrial Oxidative Metabolism in Post-Mitotic Neurons" International Journal of Molecular Sciences 15, no. 12: 22604-22625. https://doi.org/10.3390/ijms151222604