The Dynamics of DNA Methylation in Maize Roots under Pb Stress

Abstract
:
1. Introduction
2. Results
2.1. Analysis of Methylated DNA Immunoprecipitation-Sequencing (MeDIP-seq) Reads
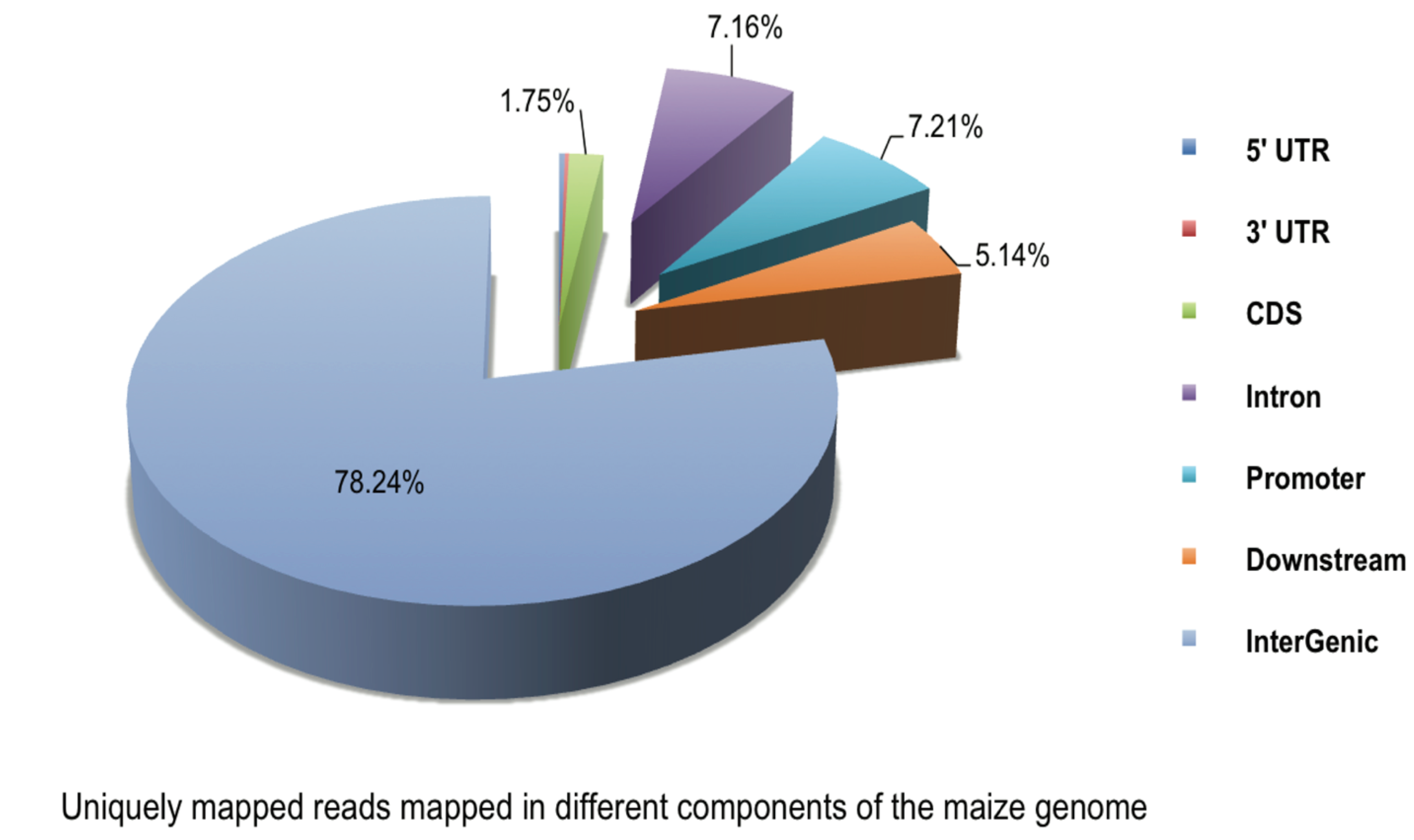
2.2. DNA Methylation Profiles of Maize Roots


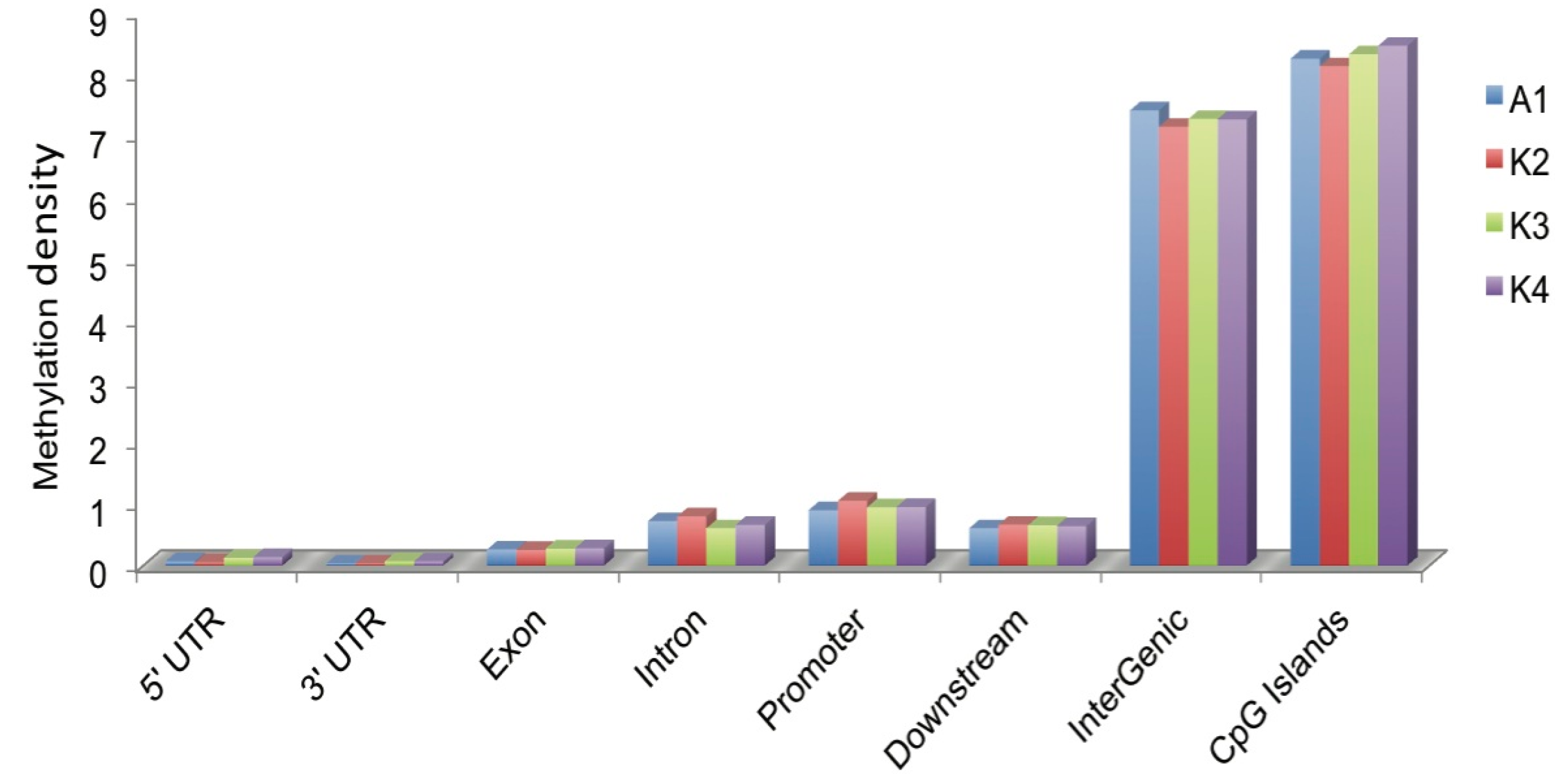
2.3. Distribution of DNA Methylation in CGIs
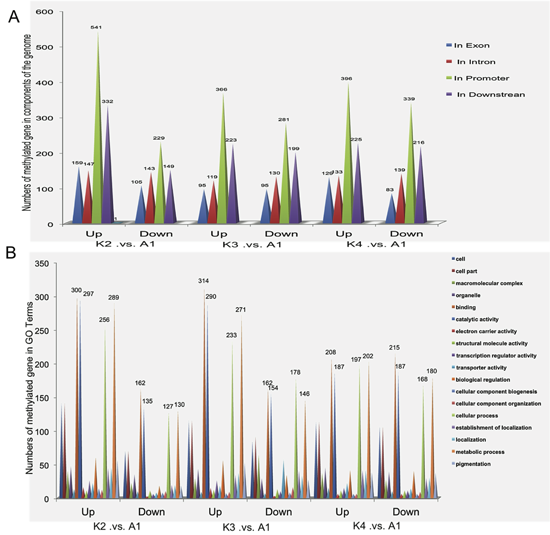
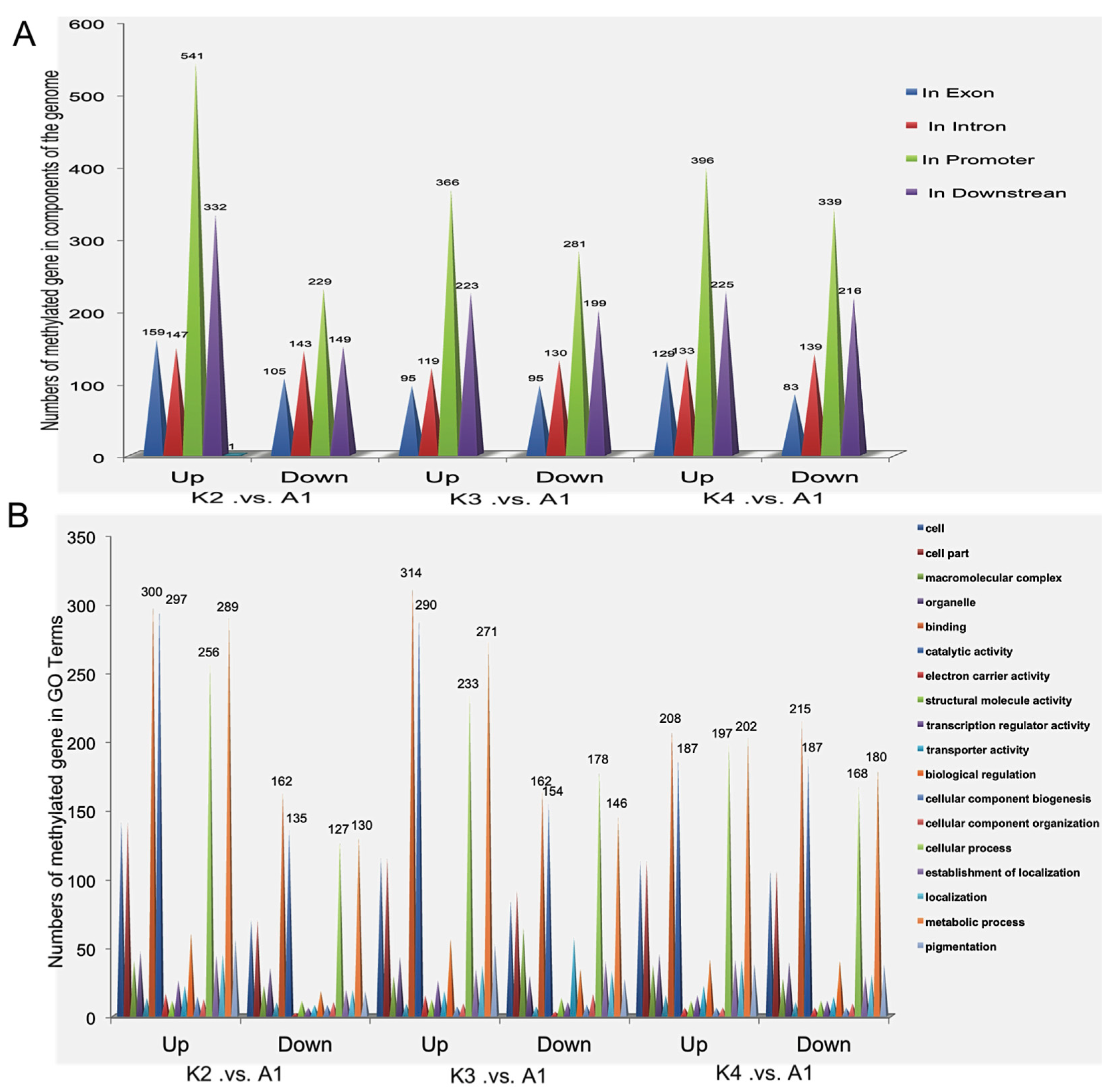
2.4. Gene Ontology (GO) Analysis of Methylated Genes in the Four Samples
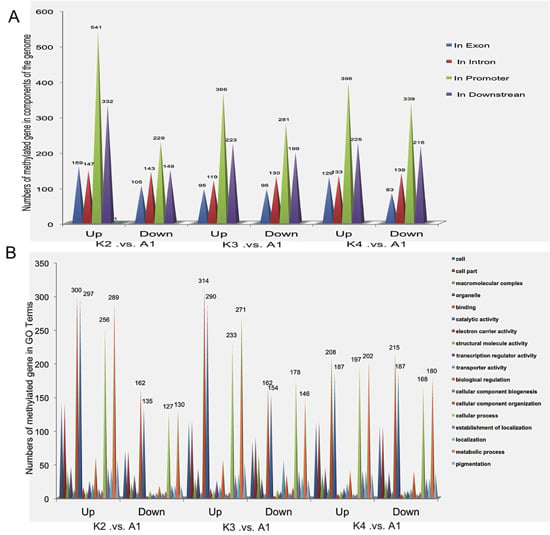
2.5. Differentially Methylated Genes among the Four Samples

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Differential Methylated Genes | K2/A1 | K3/A1 | K4/A1 | Interpro_Description | |||||
|---|---|---|---|---|---|---|---|---|---|
| GRMZM2G406099 | 8.85 | 10.41 | 7.01 | Tetratricopeptide repeat-containing domain | |||||
| GRMZM2G467695 | 0.11 | 3.54 | 0.33 | Ribosomal protein S4e, central | |||||
| GRMZM2G179910 | 0.4 | 0.46 | 0.28 | Peptidase M17, leucyl aminopeptidase, C-terminal | |||||
| GRMZM2G164705 | 2.21 | 2.47 | 3.63 | Mini-chromosome maintenance, DNA-dependent ATPase | |||||
| AC190812.3_FG006 | 0.45 | 0.56 | 0.51 | Small GTP-binding protein domain | |||||
| GRMZM2G398107 | 6.32 | 9.37 | 7.01 | Brevis radix-like domain | |||||
| GRMZM2G480171 | 2.64 | 2.84 | 2.46 | AP2/ERF domain | |||||
| GRMZM2G042133 | 0 | 0.15 | 0 | Universal stress protein A | |||||
| GRMZM2G067320 | 4.63 | 5.2 | 4.34 | Protein phosphatase inhibitor | |||||
| GRMZM2G412577 | 0.24 | 0.4 | 0.33 | Protein of unknown function DUF573 | |||||
| GRMZM2G470666 | 0.66 | 2.48 | 2.23 | Peptidyl-prolyl cis–trans isomerase, FKBP-type, domain | |||||
| GRMZM2G395348 | 0.13 | 0.21 | 0.1 | Unknown | |||||
| GRMZM2G045215 | 0 | 0.12 | 0 | Unknown | |||||
| GRMZM2G406074 | 8.85 | 10.41 | 7.01 | Zinc finger, C2H2-like | |||||
| GRMZM5G865576 | 0.14 | 2.78 | 2.29 | Zinc finger, C2H2 | |||||
| GRMZM2G065276 | 2.53 | 2.78 | 2.67 | WD4Unknown repeat | |||||
| GRMZM2G035664 | 0.38 | 0.42 | 0.35 | U box domain | |||||
| GRMZM2G010046 | 0.25 | 0.46 | 0.4 | Tify | |||||
| GRMZM2G028114 | 1.9 | 2.01 | 0 | Tetraacyldisaccharide 4'-kinase | |||||
| GRMZM2G007488 | 0 | 1.95 | 0.36 | Small GTP-binding protein domain | |||||
| GRMZM2G139882 | 0.36 | 0.48 | 0 | SANT/Myb domain | |||||
| GRMZM2G134234 | 0 | 0.15 | 0 | Protein of unknown function DUF538 | |||||
| GRMZM2G136599 | 4.42 | 4.68 | 1.6 | Unknown | |||||
| GRMZM2G379656 | 0.16 | 0.39 | 0.25 | Unknown | |||||
| GRMZM2G095239 | 0.46 | 0.55 | 0.44 | Zinc finger, RING-type | |||||
| GRMZM2G145458 | 3.48 | 5.2 | 4.75 | Glycosyl transferase, family 48 | |||||
| GRMZM2G062499 | 4.04 | 4.37 | 3.8 | F-box domain, cyclin-like | |||||
| GRMZM2G045854 | 0.45 | 0.46 | 0.31 | F-box domain, cyclin-like | |||||
| GRMZM2G057674 | 0.2 | 0.38 | 0.26 | Exocyst complex component Sec1Unknown-like | |||||
| GRMZM2G701221 | 0.19 | 0.32 | 0.15 | CDC48, N-terminal subdomain | |||||
| GRMZM2G058292 | 0.14 | 0 | 0.22 | Calponin homology domain | |||||
| GRMZM2G401869 | 0.71 | 0.64 | 0.79 | Unknown | |||||
| GRMZM2G406553 | 5.06 | 5.2 | 5.51 | Unknown | |||||
| GRMZM2G157422 | 0.3 | 0.31 | 0.27 | Unknown | |||||
| GRMZM2G020996 | 0.19 | 0.35 | 0.19 | Unknown | |||||
| GRMZM2G120298 | 0 | 0.09 | 0.33 | Uncharacterised protein family UPFUnknown261 | |||||
| GRMZM5G829337 | 0.46 | 0.52 | 0.58 | Protein of unknown function DUF76Unknown | |||||
| GRMZM2G051512 | 4.42 | 5.2 | 5.51 | Protein of unknown function DUF1644 | |||||
| GRMZM2G145721 | 0.36 | 0.45 | 0.48 | Plant regulator RWP-RK | |||||
| GRMZM2G008578 | 0.45 | 0.63 | 0.64 | 5-Formyltetrahydrofolate cyclo-ligase | |||||
| GRMZM2G315349 | 6.32 | 6.24 | 10.01 | Unknown | |||||
| GRMZM2G108418 | 0.59 | 0.6 | 0.67 | Transcriptional coactivator p15 | |||||
| AC202561.3_FG007 | 0.24 | 0.33 | 10,000 | Phosphatidyl serine synthase | |||||
| GRMZM2G121704 | 0.46 | 0.52 | 0.58 | NAD-dependent epimerase/dehydratase | |||||
| AC184857.2_FG006 | 3.16 | 3.64 | 4.5 | LURP1-like domain | |||||
| GRMZM2G145718 | 0.36 | 0.45 | 0.48 | HhH–GPD domain | |||||
| GRMZM2G049950 | 0.14 | 0.69 | 0.7 | Calcium-binding EF-hand | |||||
| GRMZM2G037941 | 0.42 | 0.46 | 0.53 | Unknown | |||||
| GRMZM2G142693 | 6.32 | 7.29 | 12.01 | Unknown | |||||
| GRMZM2G320175 | 0.42 | 0.52 | 0.58 | Pleckstrin homology domain | |||||
| GRMZM2G553532 | 0.43 | 0.46 | 0.53 | Phox-associated domain | |||||
| GRMZM2G103033 | 0.47 | 0.5 | 0.61 | 3'–5' exonuclease domain | |||||
| GRMZM2G121820 | 7.58 | 7.29 | 8.01 | Unknown | |||||
| GRMZM2G064814 | 0.37 | 0.37 | 0.42 | Unknown | |||||
| GRMZM2G026442 | 0 | 0.13 | 1.47 | Serine-threonine/tyrosine-protein kinase catalytic domain | |||||
| GRMZM2G066485 | 0.47 | 0.61 | 0.55 | SANT/Myb domain | |||||
| GRMZM2G044900 | 0.24 | 0.33 | 10,000 | Lipase, GDSL | |||||
| AC188195.2_FG004 | 0.35 | 0.35 | 0.39 | Basic-leucine zipper domain | |||||
| GRMZM2G302405 | 0.14 | 0.23 | 7.01 | GRAM | |||||
| GRMZM2G095323 | 2.11 | 2.2 | 3 | Unknown | |||||
| GRMZM2G444567 | 10,000 | 10,000 | 10,000 | K Homology domain, type 1 | |||||
| GRMZM2G433216 | 0 | 0 | 0 | Unknown | |||||
| AC213654.3_FG005 | 10,000 | 10,000 | 10,000 | Transcription factor GRAS | |||||
| GRMZM2G133129 | 0 | 0 | 0 | Domain of unknown function DUF292, eukaryotic | |||||
| GRMZM2G089596 | 10,000 | 10,000 | 10,000 | β-Lactamase-like | |||||
| GRMZM5G895991 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| GRMZM2G314946 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| GRMZM2G029055 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| GRMZM2G124524 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| GRMZM2G150866 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| GRMZM2G324886 | 10,000 | 10,000 | 10,000 | UBA-like | |||||
| GRMZM2G471931 | 10,000 | 10,000 | 10,000 | Sec1-like protein | |||||
| GRMZM5G823484 | 10,000 | 10,000 | 10,000 | Proteinase inhibitor I13, potato inhibitor I | |||||
| GRMZM5G845682 | 10,000 | 10,000 | 10,000 | Glycoside hydrolase, family 19, catalytic | |||||
| GRMZM2G080243 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| AC213654.3_FG006 | 10,000 | 10,000 | 10,000 | Ubiquitin interacting motif | |||||
| GRMZM2G031398 | 10,000 | 10,000 | 10,000 | Senescence regulator | |||||
| GRMZM2G159531 | 10,000 | 10,000 | 10,000 | Cytokinin riboside 5'-monophosphate phosphoribohydrolase LOG | |||||
| GRMZM2G025396 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| GRMZM2G017405 | 10,000 | 10,000 | 10,000 | Leucine-rich repeat-containing N-terminal, type 2 | |||||
| GRMZM2G090213 | 10,000 | 10,000 | 10,000 | FMN-dependent dehydrogenase | |||||
| GRMZM2G132464 | 10,000 | 10,000 | 10,000 | CS-like domain | |||||
| GRMZM2G056524 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| GRMZM2G071277 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| GRMZM2G525084 | 10,000 | 10,000 | 10,000 | Unknown | |||||
| AC229873.1_FG003 | 8.85 | 6.24 | 7.01 | Tetratricopeptide repeat-containing domain | |||||
| GRMZM2G316593 | 5.06 | 4.16 | 4.5 | Rab GDI protein | |||||
| AC190789.3_FG005 | 5.06 | 5.2 | 5.51 | Protein phosphatase 2C (PP2C)-like | |||||
| GRMZM2G380242 | 2.18 | 1.99 | 2.37 | Nucleic acid-binding, OB-fold | |||||
| GRMZM5G862193 | 6.32 | 6.24 | 10.01 | Bromodomain | |||||
| GRMZM2G167718 | 3.16 | 3.12 | 3.67 | Unknown | |||||
| GRMZM2G081380 | 2.76 | 2.46 | 2.46 | Unknown | |||||
| GRMZM2G078389 | 0.19 | 0.16 | 0.23 | Unknown | |||||
| GRMZM2G369243 | 0.14 | 0.12 | 0.22 | Ribosomal RNA adenine methylase transferase | |||||
| GRMZM2G148194 | 5.06 | 4.68 | 5.51 | Protein phosphatase 2C (PP2C)-like | |||||
| GRMZM2G422464 | 8.85 | 6.24 | 9.01 | Mitochondrial carrier protein | |||||
| GRMZM2G057743 | 0.49 | 0.21 | 6.51 | Kinesin, motor domain | |||||
| GRMZM2G385925 | 10,000 | 2.6 | 10,000 | CTLH, C-terminal LisH motif | |||||
| GRMZM2G097084 | 10,000 | 0.36 | 0.4 | Aminoacyl-tRNA synthetase, class 1a, anticodon-binding | |||||
| GRMZM2G401075 | 7.58 | 6.24 | 13.01 | Zinc finger, C6HC-type | |||||
| GRMZM2G447876 | 0.34 | 0.33 | 7.01 | Signal recognition particle, SRP9 subunit | |||||
| GRMZM2G427301 | 3.16 | 2.38 | 3.25 | Protein of unknown function DUF5Unknown2 | |||||
| GRMZM2G052200 | 4.11 | 3.38 | 4 | Protein of unknown function DUF1754, eukaryotic | |||||
| GRMZM2G093405 | 0.55 | 0.34 | 0.59 | Paraneoplastic encephalomyelitis antigen | |||||
| GRMZM2G392516 | 10.11 | 7.29 | 8.01 | P-type ATPase, A domain | |||||
| GRMZM2G082487 | 2.91 | 2.19 | 2.4 | Leucine-rich repeat | |||||
| GRMZM2G453296 | 3.16 | 0.52 | 0.53 | Knottin | |||||
| AC199487.4_FG002 | 2.91 | 2.39 | 2.6 | Allergen V5/Tpx-1-related | |||||
| GRMZM2G315786 | 0.37 | 0.35 | 0.38 | Zinc finger, RING-type | |||||
| GRMZM2G052880 | 10,000 | 2.08 | 10,000 | WD4Unknown repeat | |||||
| GRMZM2G434669 | 7.58 | 7.29 | 8.01 | Small-subunit processome, Utp11 | |||||
| GRMZM2G155260 | 3.07 | 2.38 | 2.57 | Ribosomal protein L2Unknown | |||||
| GRMZM2G061876 | 2.37 | 2.21 | 2.25 | Pentatricopeptide repeat | |||||
| GRMZM2G302233 | 0.37 | 0.26 | 0.42 | Pentatricopeptide repeat | |||||
| GRMZM2G121785 | 7.58 | 7.29 | 8.01 | Pectate lyase/Amb allergen | |||||
| GRMZM2G048883 | 5.06 | 4.16 | 5.51 | Leucine-rich repeat | |||||
| GRMZM2G348780 | 5.06 | 4.16 | 5.51 | Glycoside hydrolase, family 28 | |||||
| GRMZM2G035928 | 10,000 | 0.61 | 0.66 | Unknown | |||||
| GRMZM2G037627 | 7.58 | 3.47 | 4.34 | Unknown | |||||
| GRMZM2G072462 | 6.32 | 0.45 | 9.01 | Unknown | |||||
| GRMZM2G395120 | 0.44 | 0.4 | 2.25 | Protein of unknown function DUF159 | |||||
| GRMZM5G840726 | 4.04 | 4.37 | 3.8 | WD4Unknown repeat | |||||
| AC184831.3_FG003 | 10,000 | 3.12 | 3 | Kinesin, motor domain | |||||
| GRMZM2G557750 | 6.95 | 4.16 | 0 | Chaperone DnaJ, C-terminal | |||||
| AC209877.3_FG002 | 2.95 | 3.12 | 3.17 | Unknown | |||||
| GRMZM2G482657 | 5.69 | 4.16 | 2.09 | Zinc finger, RING-type | |||||
| GRMZM2G003595 | 10,000 | 10,000 | 1.88 | Zinc finger, LSD1-type | |||||
| GRMZM2G075096 | 10,000 | 4.68 | 4.5 | ATPase, AAA-type, conserved site | |||||
| GRMZM2G001904 | 2.17 | 2.03 | 2 | Adenylosuccinate lyase | |||||
| GRMZM2G040079 | 6.95 | 5.72 | 5 | Unknown | |||||
| GRMZM2G011932 | 5.06 | 4.68 | 4.5 | Unknown | |||||
| GRMZM2G152853 | 3.48 | 3.38 | 3 | START domain | |||||
| GRMZM2G702889 | 15.17 | 9.37 | 8.01 | Proteinase inhibitor I13, potato inhibitor I | |||||
| GRMZM2G065205 | 10.11 | 9.37 | 9.01 | Unknown | |||||
| GRMZM2G138410 | 10,000 | 10,000 | 0.22 | Zinc finger, RING-type | |||||
| GRMZM2G040164 | 6.95 | 5.72 | 5 | SANT/Myb domain | |||||
| GRMZM2G435373 | 4.42 | 3.82 | 0.22 | Unknown | |||||
| GRMZM2G339009 | 10,000 | 10,000 | 0.14 | Unknown | |||||
| GRMZM2G348726 | 10.11 | 6.24 | 2.56 | Proteasome, subunit α/β | |||||
| GRMZM2G133958 | 10,000 | 3.12 | 3 | NUDIX hydrolase domain |

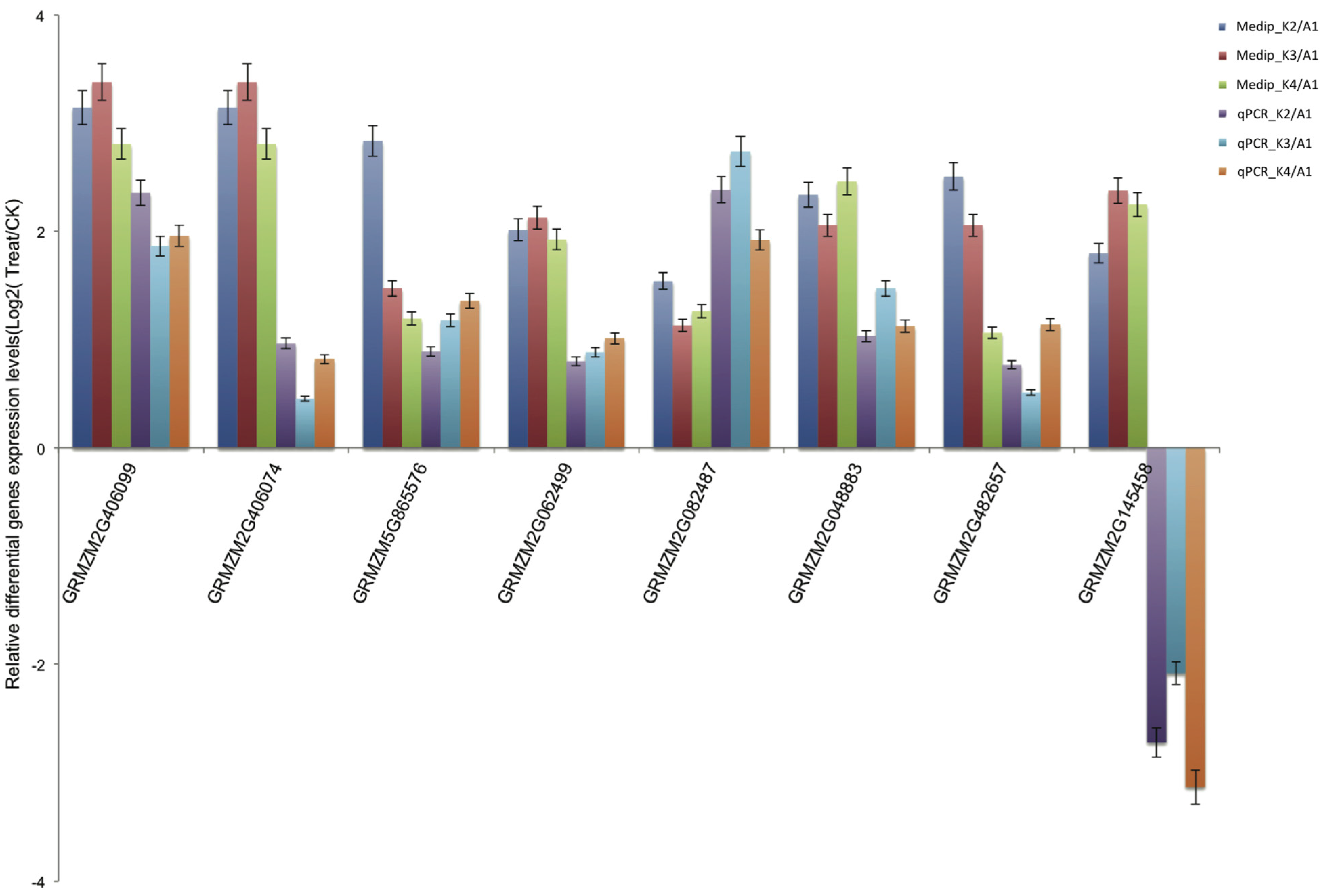
2.6. Validation of Differentially Methylated Genes by Quantitative Real-Time PCR (qRT-PCR)

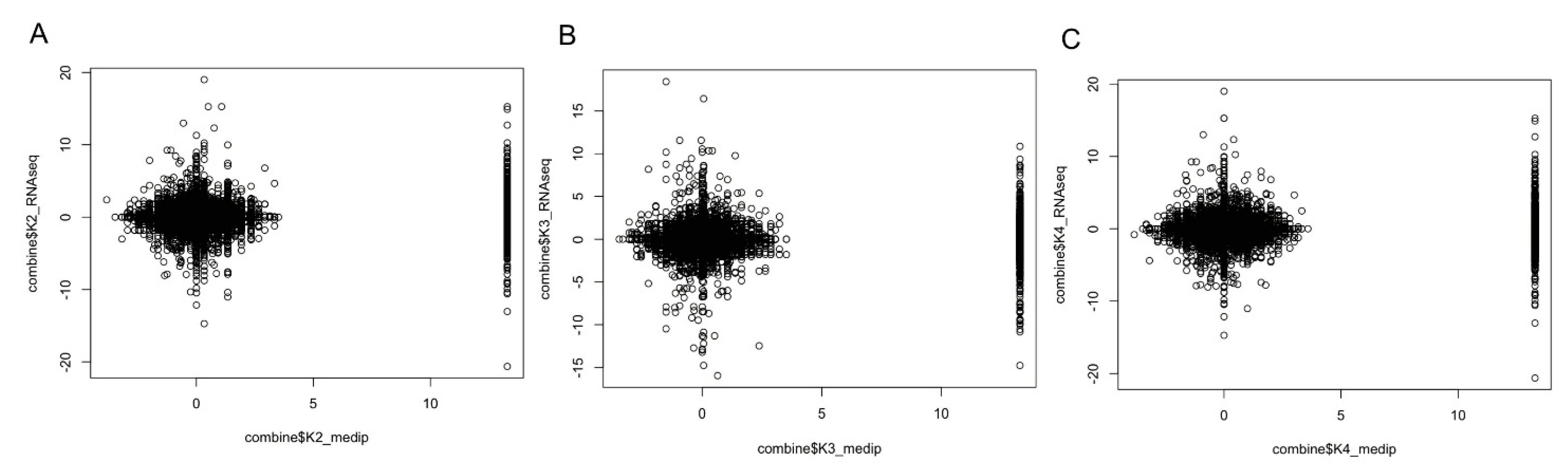
2.7. Promoter DNA Methylation and Gene Expression Level

3. Discussion
3.1. DNA Methylation Profiles
3.2. Functions of Genes Potentially Methylated in Response to Pb Stress
4. Experimental Section
4.1. Seed Sterilization and Experiment Design
4.2. DNA Extraction and Preparation for MeDIP-seq
4.3. Bioinformatic Analysis
4.4. Reverse Transcription, Standard and Real-Time Reverse Transcription PCR
4.5. GO Annotation of All Genes with Peaks
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Abbreviations
| CGIs | CpG islands |
| AP2/ERF | APETALA2/ethylene responsive factor |
| bHLH | basic helix–loop–helix |
| MYB | myeloblastosis |
| bZIP | basic leucine zipper domain |
| GRAS | GAI, RGA, SCR |
| MeDIP-seq | methylated DNA immunoprecipitation-sequencing |
Conflicts of Interest
References
- Piechalak, A.; Tomaszewska, B.; Baralkiewicz, D.; Malecka, A. Accumulation and detoxification of lead ions in legumes. Phytochemistry 2002, 60, 153–162. [Google Scholar]
- Verma, S.; Dubey, R. Lead toxicity induces lipid peroxidation and alters the activities of antioxidant enzymes in growing rice plants. Plant Sci. 2003, 164, 645–655. [Google Scholar]
- Nocito, F.F.; Lancilli, C.; Crema, B.; Fourcroy, P.; Davidian, J.-C.; Sacchi, G.A. Heavy metal stress and sulfate uptake in maize roots. Plant Physiol. 2006, 141, 1138–1148. [Google Scholar]
- Gupta, D.; Huang, H.; Corpas, F. Lead tolerance in plants: Strategies for phytoremediation. Environ. Sci. Pollut. Res. 2013, 20, 2150–2161. [Google Scholar]
- Zhang, Z.; Jin, F.; Wang, C.; Luo, J.; Lin, H.; Xiang, K.; Liu, L.; Zhao, M.; Zhang, Y.; Ding, H. Difference between Pb and Cd accumulation in 19 elite maize inbred lines and application prospects. BioMed Res. Int. 2012, 2012, 271485. [Google Scholar]
- Shen, Y.; Zhang, Y.; Chen, J.; Lin, H.; Zhao, M.; Peng, H.; Liu, L.; Yuan, G.; Zhang, S.; Zhang, Z. Genome expression profile analysis reveals important transcripts in maize roots responding to the stress of heavy metal Pb. Physiol. Plant. 2013, 147, 270–282. [Google Scholar]
- Chinnusamy, V.; Zhu, J.-K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar]
- Labra, M.; Ghiani, A.; Citterio, S.; Sgorbati, S.; Sala, F.; Vannini, C.; Ruffini-Castiglione, M.; Bracale, M. Analysis of cytosine methylation pattern in response to water deficit in pea root tips. Plant Biol. 2002, 4, 694–699. [Google Scholar]
- Zhong, L.; Xu, Y.-H.; Wang, J.-B. DNA-methylation changes induced by salt stress in wheat Triticum aestivum. Afr. J. Biotechnol. 2009, 8, 6201–6207. [Google Scholar]
- Peng, H.; Zhang, J. Plant genomic DNA methylation in response to stresses: Potential applications and challenges in plant breeding. Prog. Nat. Sci. 2009, 19, 1037–1045. [Google Scholar]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. 2012, 109, E2183–E2191. [Google Scholar]
- Henderson, I.R.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar]
- Eichten, S.R.; Briskine, R.; Song, J.; Li, Q.; Swanson-Wagner, R.; Hermanson, P.J.; Waters, A.J.; Starr, E.; West, P.T.; Tiffin, P. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell Online 2013, 25, 2783–2797. [Google Scholar]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar]
- Zhang, X.; Shiu, S.; Cal, A.; Borevitz, J.O. Global analysis of genetic, epigenetic and transcriptional polymorphisms in Arabidopsis thaliana using whole genome tiling arrays. PLoS Genet. 2008, 4, e1000032. [Google Scholar]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar]
- Ma, H.-S.; Liang, D.; Shuai, P.; Xia, X.-L.; Yin, W.-L. The salt-and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J. Exp. Bot. 2010, 61, 4011–4019. [Google Scholar]
- Knight, H.; Knight, M.R. Abiotic stress signalling pathways: Specificity and cross-talk. Trends Plant Sci. 2001, 6, 262–267. [Google Scholar]
- Leung, J.; Merlot, S.; Giraudat, J. The Arabidopsis abscisic acid-insensitive2 (ABI2) and ABI1 genes encode homologous protein phosphatases 2C involved in abscisic acid signal transduction. Plant Cell Online 1997, 9, 759–771. [Google Scholar]
- Sun, H.-L.; Wang, X.-J.; Ding, W.-H.; Zhu, S.-Y.; Zhao, R.; Zhang, Y.-X.; Xin, Q.; Wang, X.-F.; Zhang, D.-P. Identification of an important site for function of the type 2C protein phosphatase ABI2 in abscisic acid signalling in Arabidopsis. J. Exp. Bot. 2011, 62, 5713–5725. [Google Scholar]
- Azevedo, C.; Santos-Rosa, M.J.; Shirasu, K. The U-box protein family in plants. Trends Plant Sci. 2001, 6, 354–358. [Google Scholar]
- Zhou, Z.S.; Yang, S.N.; Li, H.; Zhu, C.C.; Liu, Z.P.; Yang, Z.M. Molecular dissection of mercury-responsive transcriptome and sense/antisense genes in Medicago truncatula. J. Hazard. Mater. 2013, 252, 123–131. [Google Scholar]
- Zheng, J.; Fu, J.; Gou, M.; Huai, J.; Liu, Y.; Jian, M.; Huang, Q.; Guo, X.; Dong, Z.; Wang, H. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol. Biol. 2010, 72, 407–421. [Google Scholar]
- Lakhssassi, N.; Doblas, V.G.; Rosado, A.; del Valle, A.E.; Posé, D.; Jimenez, A.J.; Castillo, A.G.; Valpuesta, V.; Borsani, O.; Botella, M.A. The Arabidopsis tetratricopeptide thioredoxin-like gene family is required for osmotic stress tolerance and male sporogenesis. Plant Physiol. 2012, 158, 1252–1266. [Google Scholar]
- Rosado, A.; Schapire, A.L.; Bressan, R.A.; Harfouche, A.L.; Hasegawa, P.M.; Valpuesta, V.; Botella, M.A. The Arabidopsis tetratricopeptide repeat-containing protein TTL1 is required for osmotic stress responses and abscisic acid sensitivity. Plant Physiol. 2006, 142, 1113–1126. [Google Scholar]
- Lemaire, K.; Moura, R.F.; Granvik, M.; Igoillo-Esteve, M.; Hohmeier, H.E.; Hendrickx, N.; Newgard, C.B.; Waelkens, E.; Cnop, M.; Schuit, F. Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS One 2011, 6, e18517. [Google Scholar]
- Xu, S.-L.; Rahman, A.; Baskin, T.I.; Kieber, J.J. Two leucine-rich repeat receptor kinases mediate signaling, linking cell wall biosynthesis and ACC synthase in Arabidopsis. Plant Cell Online 2008, 20, 3065–3079. [Google Scholar]
- Durand, T.C.; Sergeant, K.; Planchon, S.; Carpin, S.; Label, P.; Morabito, D.; Hausman, J.F.; Renaut, J. Acute metal stress in Populus tremula×P. alba (717-1B4 genotype): Leaf and cambial proteome changes induced by cadmium2+. Proteomics 2010, 10, 349–368. [Google Scholar]
- Gramene. Avaible online: http://ensembl.gramene.org/Zea_mays/Info/Index (accessed on 1 August 2014).
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar]
- EMBOSS Cpgplot. Available online: http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/ (accessed on 1 June 2000).
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar]
- Model-Based Analysis for ChIP-Seq (MACS). Avaible online: http://liulab.dfci.harvard.edu/MACS/ (accessed on 19 June 2011).
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, H.; Gao, J.; Qin, C.; Ma, H.; Huang, H.; Song, P.; Luo, X.; Lin, H.; Shen, Y.; Pan, G.; et al. The Dynamics of DNA Methylation in Maize Roots under Pb Stress. Int. J. Mol. Sci. 2014, 15, 23537-23554. https://doi.org/10.3390/ijms151223537
Ding H, Gao J, Qin C, Ma H, Huang H, Song P, Luo X, Lin H, Shen Y, Pan G, et al. The Dynamics of DNA Methylation in Maize Roots under Pb Stress. International Journal of Molecular Sciences. 2014; 15(12):23537-23554. https://doi.org/10.3390/ijms151223537
Chicago/Turabian StyleDing, Haiping, Jian Gao, Cheng Qin, Haixia Ma, Hong Huang, Pan Song, Xirong Luo, Haijian Lin, Ya'ou Shen, Guangtang Pan, and et al. 2014. "The Dynamics of DNA Methylation in Maize Roots under Pb Stress" International Journal of Molecular Sciences 15, no. 12: 23537-23554. https://doi.org/10.3390/ijms151223537