Generation of Bladder Urothelium from Human Pluripotent Stem Cells under Chemically Defined Serum- and Feeder-Free System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
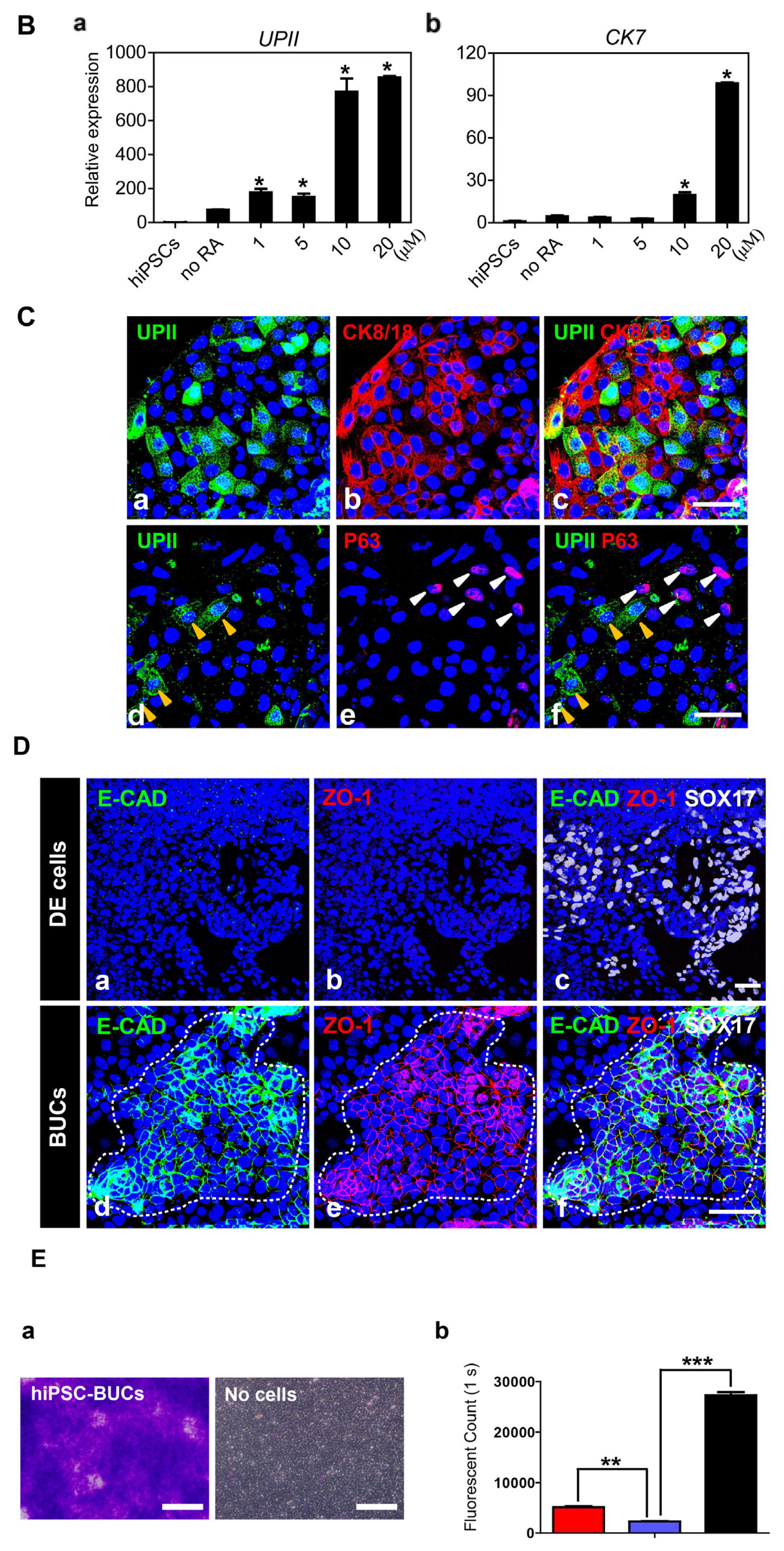
2.1.1. Induction of Definitive Endoderm from hESCs
2.1.2. Specification of hESC-Derived DE Cells into BUCs
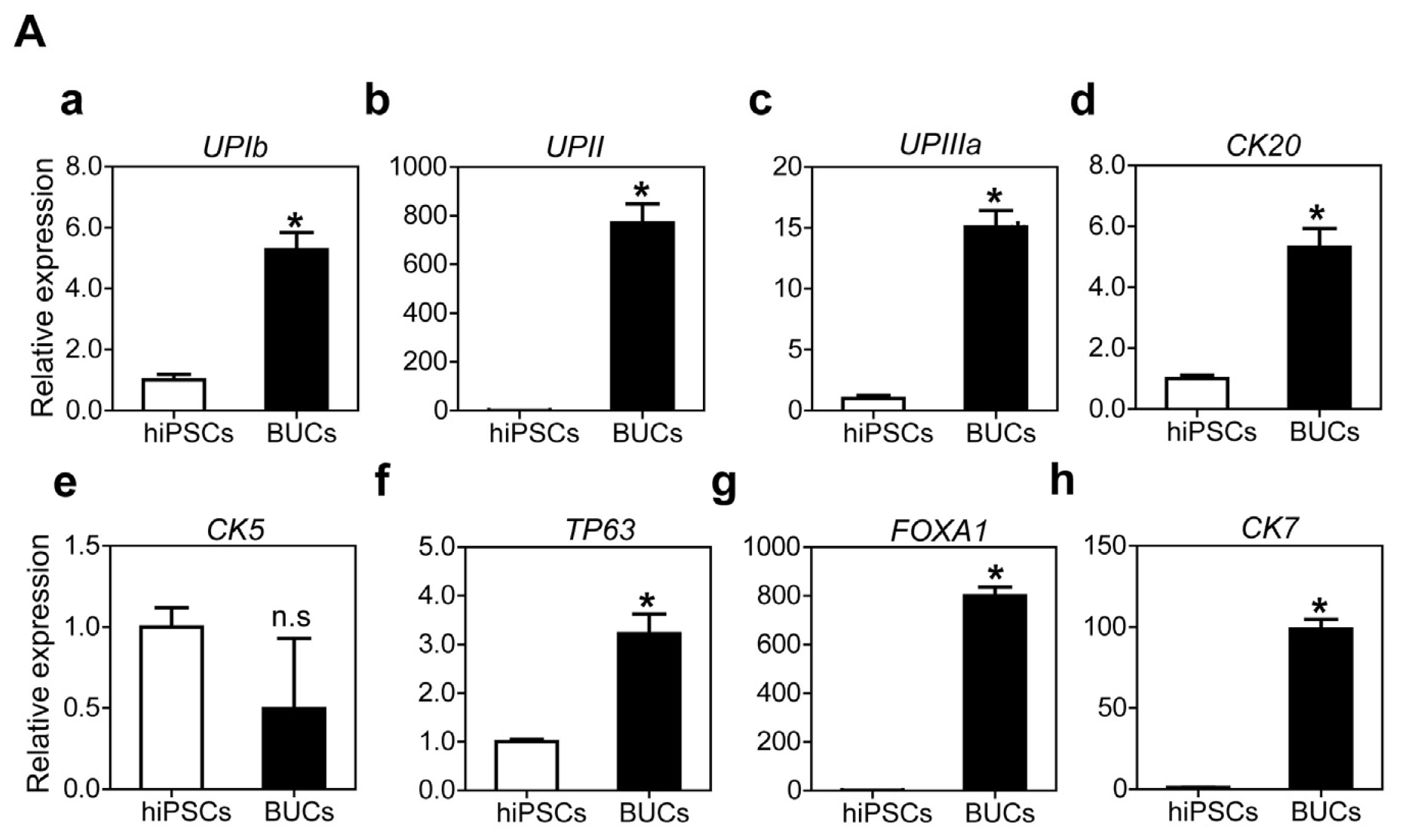
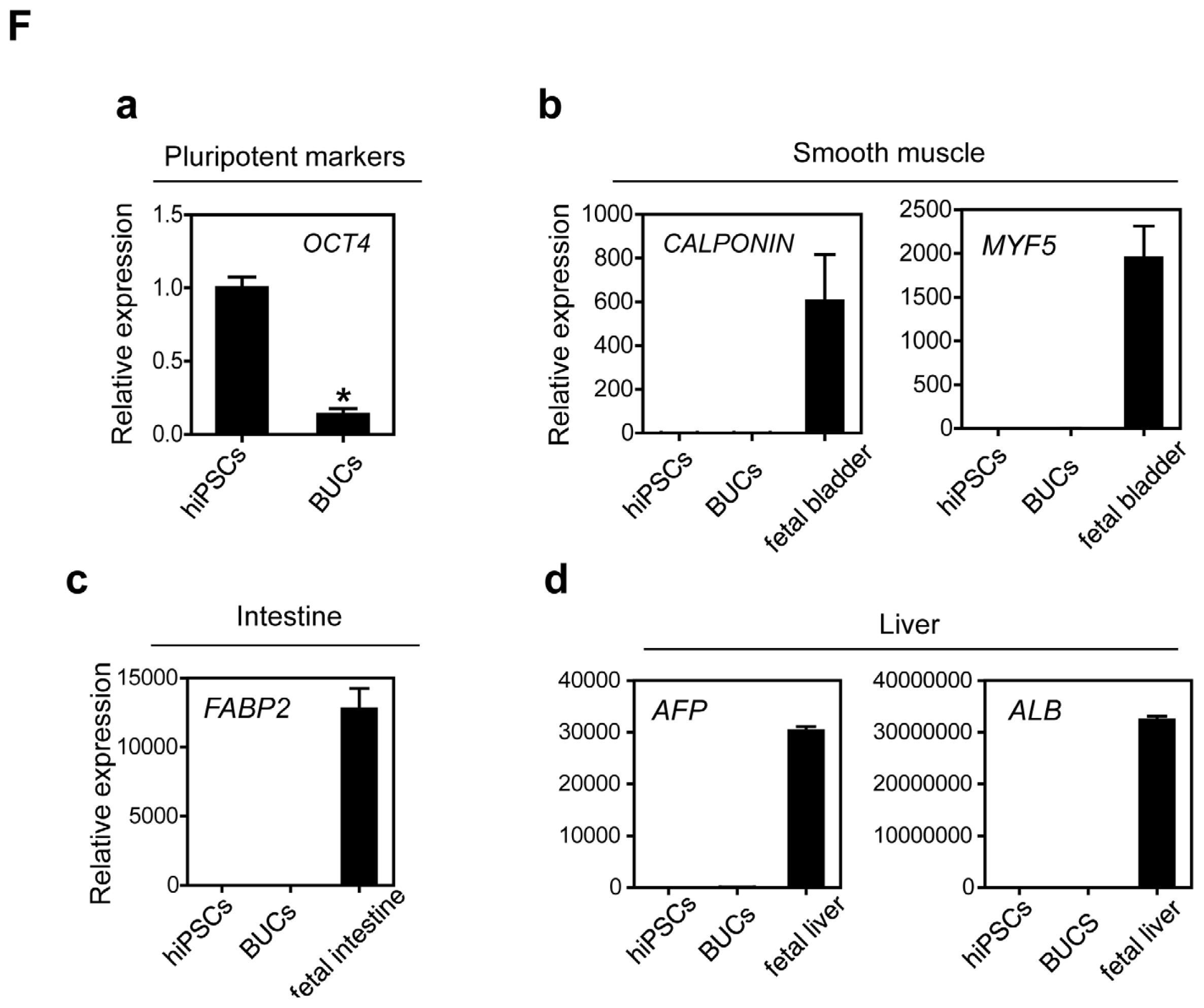
2.1.3. Sequential Differentiation of hiPSCs into DE and BUCs
2.2. Discussion
3. Experimental Section
3.1. Generation of hiPSCs Derived from CRL-2097 Fibroblasts
3.2. Maintenance of Human Pluripotent Stem Cells
3.3. Differentiation of BUCs from hPSCs
3.4. RNA Preparation and Real Time RT-PCR
3.5. Immunocytochemistry
3.6. Flow Cytometric Analysis
3.7. In Vitro Permeability Assay
3.8. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-15-07139-s001.pdfAcknowledgments
Conflicts of Interest
Abbreviations
| hPSCs | Human pluripotent stem cells |
| hESCs | Human embryonic stem cells |
| hiPSCs | Human induced pluripotent stem cells |
| DE | Definitive endoderm |
| BUCs | Bladder urothelial cells |
| UP | UROPLAKIN |
| CK | CYTOKERATIN |
| AW | Activin and Wnt3a |
| RA | Retinoic acid |
| K-SFM | Keratinocyte-specific serum-free medium |
- Author ContributionsStudy conception and design: Minyong Kang and Hyeon Hoe Kim; Acquisition of data: Minyong Kang; Analysis and interpretation of data: Minyong Kang and Yong-Mahn Han; Drafting the manuscript: Minyong Kang and Yong-Mahn Han; Critical revision: Minyong Kang, Hyeon Hoe Kim and Yong-Mahn Han.
References
- Sharma, A.K. An examination of regenerative medicine-based strategies for the urinary bladder. Regen. Med 2011, 6, 583–598. [Google Scholar]
- Biers, S.M.; Venn, S.N.; Greenwell, T.J. The past, present and future of augmentation cystoplasty. BJU Int 2012, 109, 1280–1293. [Google Scholar]
- Petrovic, V.; Stankovic, J.; Stefanovic, V. Tissue engineering of the urinary bladder: Current concepts and future perspectives. Sci. World J 2011, 11, 1479–1488. [Google Scholar]
- Hautmann, R.E. Urinary diversion: Ileal conduit to neobladder. J. Urol 2003, 169, 834–842. [Google Scholar]
- Atala, A.; Bauer, S.B.; Hendren, W.H.; Retik, A.B. The effect of gastric augmentation on bladder function. J. Urol 1993, 149, 1099–1102. [Google Scholar]
- Drewa, T.; Adamowicz, J.; Sharma, A. Tissue engineering for the oncologic urinary bladder. Nat. Rev. Urol 2012, 9, 561–572. [Google Scholar]
- Birder, L.A.; Ruggieri, M.; Takeda, M.; van Koeveringe, G.; Veltkamp, S.; Korstanje, C.; Parsons, B.; Fry, C.H. How does the urothelium affect bladder function in health and disease?: ICI-RS 2011. Neurourol. Urodynam 2012, 31, 293–299. [Google Scholar]
- Koh, C.J.; Atala, A. Tissue engineering, stem cells, and cloning: Opportunities for regenerative medicine. J. Am. Soc. Nephrol 2004, 15, 1113–1125. [Google Scholar]
- Kim, J.H.; Lee, S.R.; Song, Y.S.; Lee, H.J. Stem cell therapy in bladder dysfunction: Where are we? And where do we have to go? BioMed Res. Int 2013, 2013, 930713. [Google Scholar]
- Kuehnle, I.; Goodell, M.A. The therapeutic potential of stem cells from adults. BMJ 2002, 325, 372–376. [Google Scholar]
- Strauer, B.E.; Kornowski, R. Stem cell therapy in perspective. Circulation 2003, 107, 929–934. [Google Scholar]
- Baraniak, P.R.; McDevitt, T.C. Stem cell paracrine actions and tissue regeneration. Regen. Med 2010, 5, 121–143. [Google Scholar]
- Okano, H.; Nakamura, M.; Yoshida, K.; Okada, Y.; Tsuji, O.; Nori, S.; Ikeda, E.; Yamanaka, S.; Miura, K. Steps toward safe cell therapy using induced pluripotent stem cells. Circ. Res 2013, 112, 523–533. [Google Scholar]
- Lebkowski, J.S.; Gold, J.; Xu, C.; Funk, W.; Chiu, C.P.; Carpenter, M.K. Human embryonic stem cells: Culture, differentiation, and genetic modification for regenerative medicine applications. Cancer J 2001, 7 Suppl 2, S83–S93. [Google Scholar]
- Mauney, J.R.; Ramachandran, A.; Yu, R.N.; Daley, G.Q.; Adam, R.M.; Estrada, C.R. All-trans retinoic acid directs urothelial specification of murine embryonic stem cells via GATA4/6 signaling mechanisms. PLos One 2010, 5, e11513. [Google Scholar]
- Oottamasathien, S.; Wang, Y.; Williams, K.; Franco, O.E.; Wills, M.L.; Thomas, J.C.; Saba, K.; Sharif-Afshar, A.R.; Makari, J.H.; Bhowmick, N.A.; et al. Directed differentiation of embryonic stem cells into bladder tissue. Dev. Biol 2007, 304, 556–566. [Google Scholar]
- Kinebuchi, Y.; Johkura, K.; Sasaki, K.; Imamura, T.; Mimura, Y.; Nishizawa, O. Direct induction of layered tissues from mouse embryonic stem cells: Potential for differentiation into urinary tract tissue. Cell Tissue Res 2008, 331, 605–615. [Google Scholar]
- Moad, M.; Pal, D.; Hepburn, A.C.; Williamson, S.C.; Wilson, L.; Lako, M.; Armstrong, L.; Hayward, S.W.; Franco, O.E.; Cates, J.M.; et al. A novel model of urinary tract differentiation, tissue regeneration, and disease: Reprogramming human prostate and bladder cells into induced pluripotent stem cells. Eur. Urol 2013, 64, 753–761. [Google Scholar]
- Southgate, J.; Hutton, K.A.; Thomas, D.F.; Trejdosiewicz, L.K. Normal human urothelial cells in vitro: Proliferation and induction of stratification. Lab. Investig 1994, 71, 583–594. [Google Scholar]
- Liu, J.; Huang, J.; Lin, T.; Zhang, C.; Yin, X. Cell-to-cell contact induces human adipose tissue-derived stromal cells to differentiate into urothelium-like cells in vitro. Biochem. Biophys. Res. Commun. 2009, 390, 931–936. [Google Scholar]
- Chung, S.S.; Koh, C.J. Bladder cancer cell in co-culture induces human stem cell differentiation to urothelial cells through paracrine FGF10 signaling. In Vitro Cell. Dev. Biol. Anim 2013, 49, 746–751. [Google Scholar]
- Zhang, M.; Peng, Y.; Zhou, Z.; Zhou, J.; Wang, Z.; Lu, M. Differentiation of human adipose-derived stem cells co-cultured with urothelium cell line toward a urothelium-like phenotype in a nude murine model. Urology 2013, 81, 465.e15–22. [Google Scholar]
- Yamada, G.; Satoh, Y.; Baskin, L.S.; Cunha, G.R. Cellular and molecular mechanisms of development of the external genitalia. Differ. Res. Biol. Divers 2003, 71, 445–460. [Google Scholar]
- Erman, A.; Veranic, P.; Psenicnik, M.; Jezernik, K. Superficial cell differentiation during embryonic and postnatal development of mouse urothelium. Tissue Cell 2006, 38, 293–301. [Google Scholar]
- Yamada, G.; Suzuki, K.; Haraguchi, R.; Miyagawa, S.; Satoh, Y.; Kamimura, M.; Nakagata, N.; Kataoka, H.; Kuroiwa, A.; Chen, Y. Molecular genetic cascades for external genitalia formation: An emerging organogenesis program. Dev. Dyn 2006, 235, 1738–1752. [Google Scholar]
- Abler, L.L.; Keil, K.P.; Mehta, V.; Joshi, P.S.; Schmitz, C.T.; Vezina, C.M. A high-resolution molecular atlas of the fetal mouse lower urogenital tract. Dev. Dyn 2011, 240, 2364–2377. [Google Scholar]
- Baker, L.A.; Gomez, R.A. Embryonic development of the ureter. Semin. Nephrol 1998, 18, 569–584. [Google Scholar]
- Staack, A.; Hayward, S.W.; Baskin, L.S.; Cunha, G.R. Molecular, cellular and developmental biology of urothelium as a basis of bladder regeneration. Differentiation 2005, 73, 121–133. [Google Scholar]
- Bilic, J.; Izpisua Belmonte, J.C. Concise review: Induced pluripotent stem cells versus embryonic stem cells: Close enough or yet too far apart? Stem Cells 2012, 30, 33–41. [Google Scholar]
- Puri, M.C.; Nagy, A. Concise review: Embryonic stem cells versus induced pluripotent stem cells: The game is on. Stem Cells 2012, 30, 10–14. [Google Scholar]
- Nagele, U.; Maurer, S.; Feil, G.; Bock, C.; Krug, J.; Sievert, K.D.; Stenzl, A. In vitro investigations of tissue-engineered multilayered urothelium established from bladder washings. Eur. Urol 2008, 54, 1414–1422. [Google Scholar]
- Feil, G.; Maurer, S.; Nagele, U.; Krug, J.; Bock, C.; Sievert, K.D.; Stenzl, A. Immunoreactivity of p63 in monolayered and in vitro stratified human urothelial cell cultures compared with native urothelial tissue. Eur. Urol 2008, 53, 1066–1073. [Google Scholar]
- Sangha, N. Isolation of urothelial cells from bladder tissue. Methods Mol. Biol 2013, 1001, 21–33. [Google Scholar]
- Bayha, E.; Jorgensen, M.C.; Serup, P.; Grapin-Botton, A. Retinoic acid signaling organizes endodermal organ specification along the entire antero-posterior axis. PLoS One 2009, 4, e5845. [Google Scholar]
- Liang, F.X.; Bosland, M.C.; Huang, H.; Romih, R.; Baptiste, S.; Deng, F.M.; Wu, X.R.; Shapiro, E.; Sun, T.T. Cellular basis of urothelial squamous metaplasia: Roles of lineage heterogeneity and cell replacement. J. Cell Biol 2005, 171, 835–844. [Google Scholar]
- Gandhi, D.; Molotkov, A.; Batourina, E.; Schneider, K.; Dan, H.; Reiley, M.; Laufer, E.; Metzger, D.; Liang, F.; Liao, Y.; et al. Retinoid signaling in progenitors controls specification and regeneration of the urothelium. Dev. Cell 2013, 26, 469–482. [Google Scholar]
- Park, S.W.; Jun Koh, Y.; Jeon, J.; Cho, Y.H.; Jang, M.J.; Kang, Y.; Kim, M.J.; Choi, C.; Sook Cho, Y.; Chung, H.M.; et al. Efficient differentiation of human pluripotent stem cells into functional CD34+ progenitor cells by combined modulation of the MEK/ERK and BMP4 signaling pathways. Blood 2010, 116, 5762–5772. [Google Scholar]
- Agarwal, S.; Holton, K.L.; Lanza, R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells 2008, 26, 1117–1127. [Google Scholar]
- Uosaki, H.; Fukushima, H.; Takeuchi, A.; Matsuoka, S.; Nakatsuji, N.; Yamanaka, S.; Yamashita, J.K. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLoS One 2011, 6, e23657. [Google Scholar]
- Hu, B.Y.; Weick, J.P.; Yu, J.; Ma, L.X.; Zhang, X.Q.; Thomson, J.A.; Zhang, S.C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar]









© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kang, M.; Kim, H.H.; Han, Y.-M. Generation of Bladder Urothelium from Human Pluripotent Stem Cells under Chemically Defined Serum- and Feeder-Free System. Int. J. Mol. Sci. 2014, 15, 7139-7157. https://doi.org/10.3390/ijms15057139
Kang M, Kim HH, Han Y-M. Generation of Bladder Urothelium from Human Pluripotent Stem Cells under Chemically Defined Serum- and Feeder-Free System. International Journal of Molecular Sciences. 2014; 15(5):7139-7157. https://doi.org/10.3390/ijms15057139
Chicago/Turabian StyleKang, Minyong, Hyeon Hoe Kim, and Yong-Mahn Han. 2014. "Generation of Bladder Urothelium from Human Pluripotent Stem Cells under Chemically Defined Serum- and Feeder-Free System" International Journal of Molecular Sciences 15, no. 5: 7139-7157. https://doi.org/10.3390/ijms15057139