Huperzine A Ameliorates Cognitive Deficits in Streptozotocin-Induced Diabetic Rats

Abstract
:1. Introduction
2. Results
2.1. Effects of HupA on Body Weight and Blood Glucose Levels
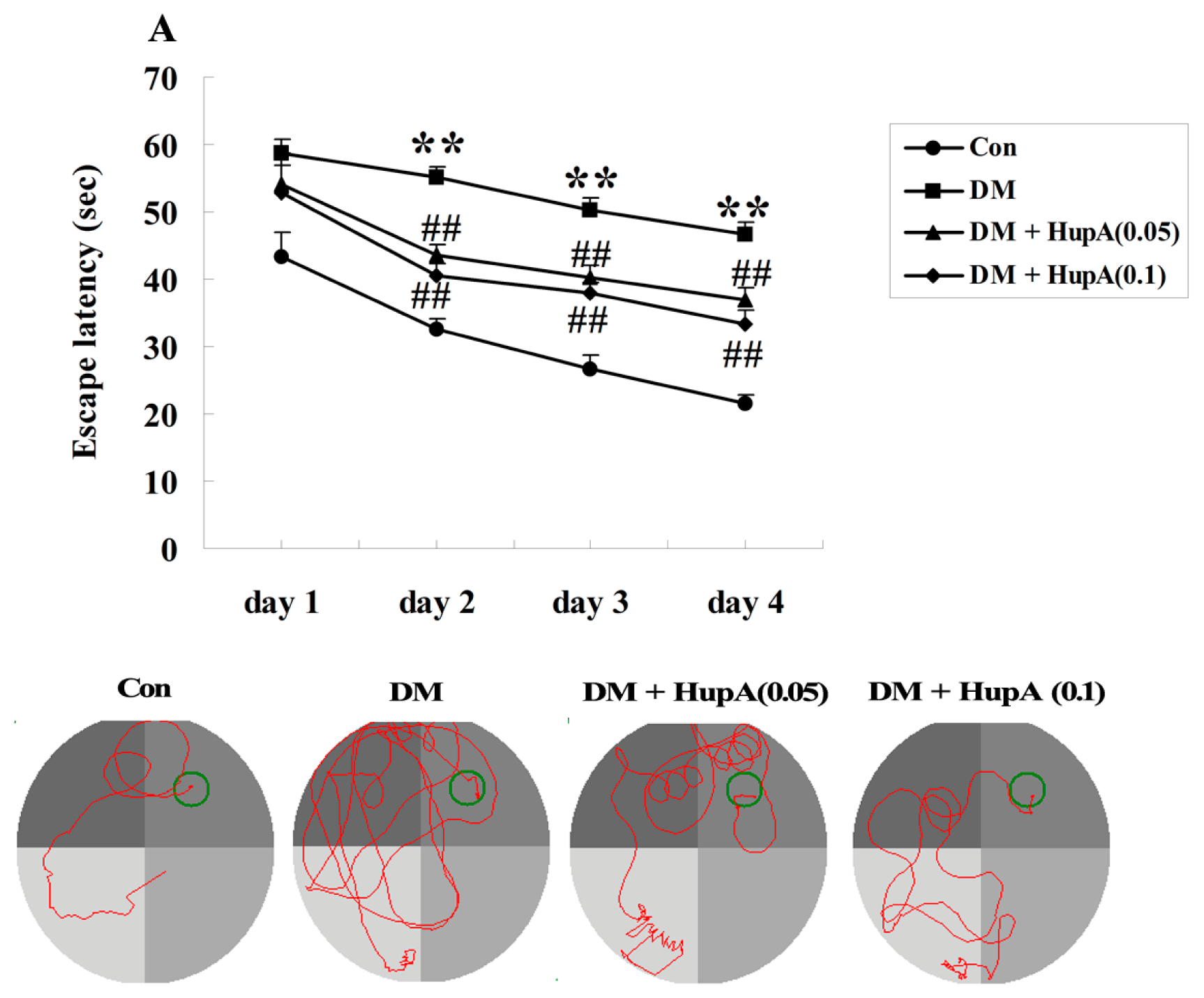
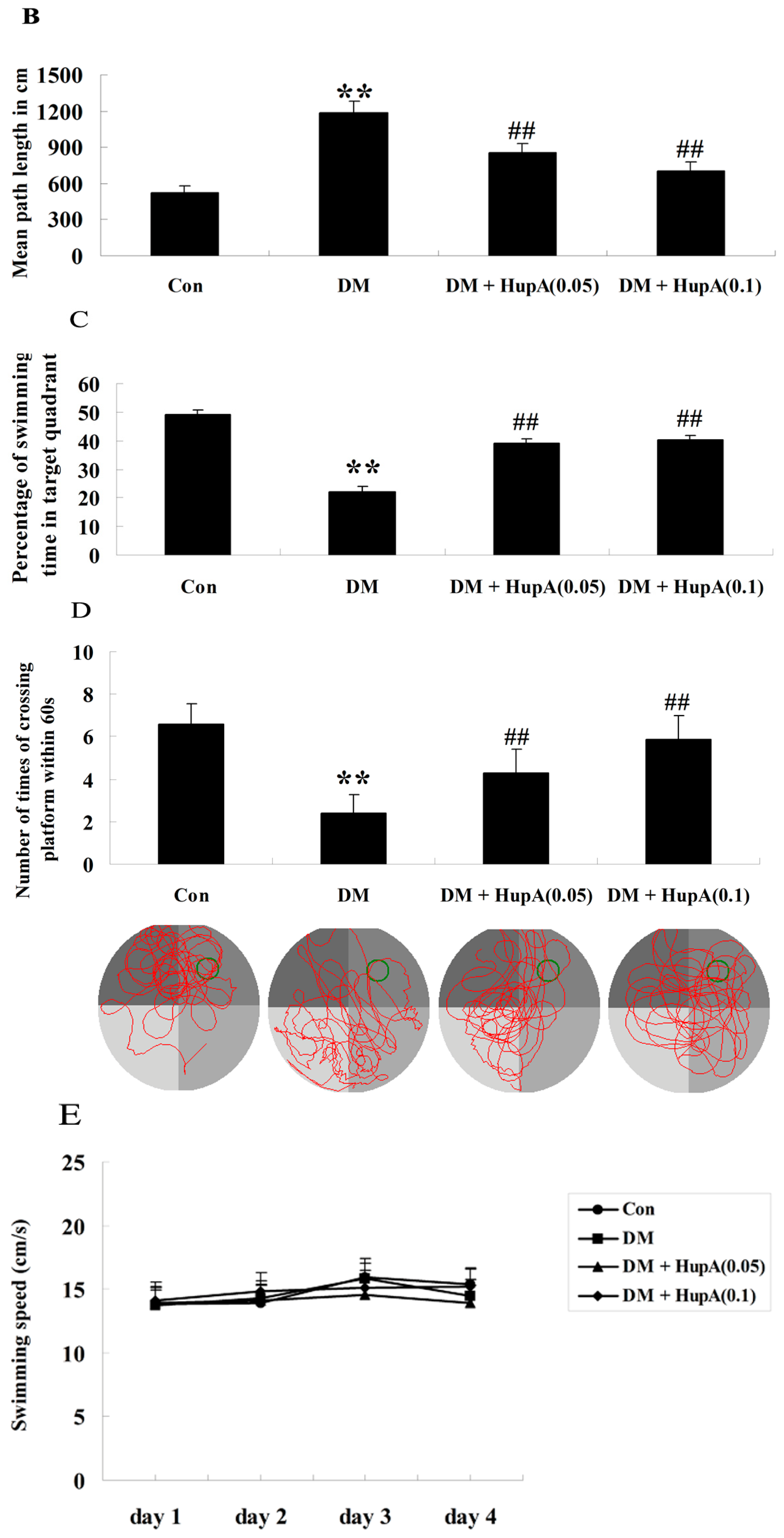
2.2. Effects of HupA on Diabetes-Induced Cognitive Deficit
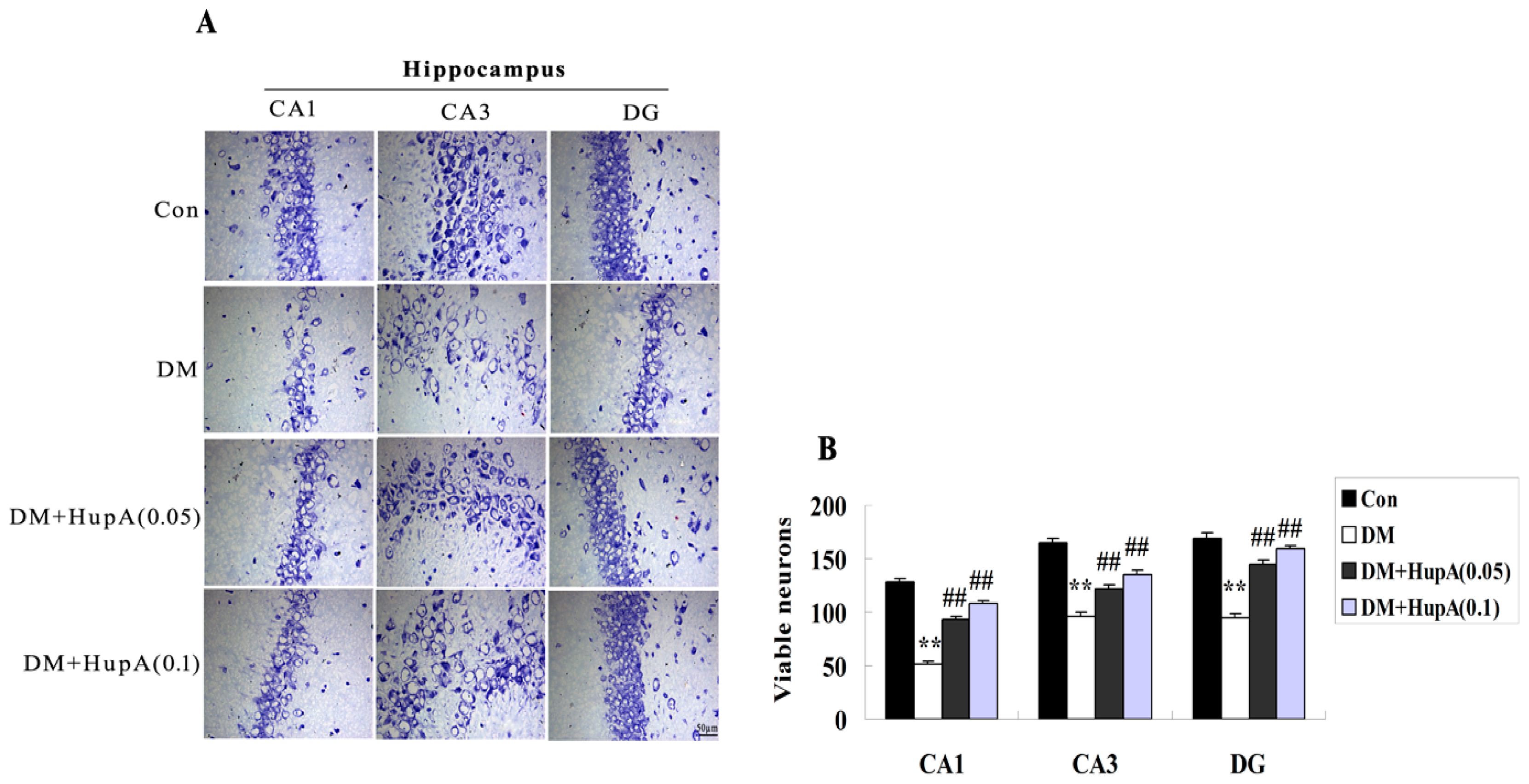
2.3. Effects of HupA on the Hippocampal Neuronal Loss in Diabetic Rats
2.4. Effects of HupA on the Activities of AChE and ChAT in Diabetic Rats Brain
2.5. Effects of HupA on mRNA and Protein Levels of BDNF in Diabetic Rat Brain
2.6. Effects of HupA on Oxidative Stress in Diabetic Rat Brain
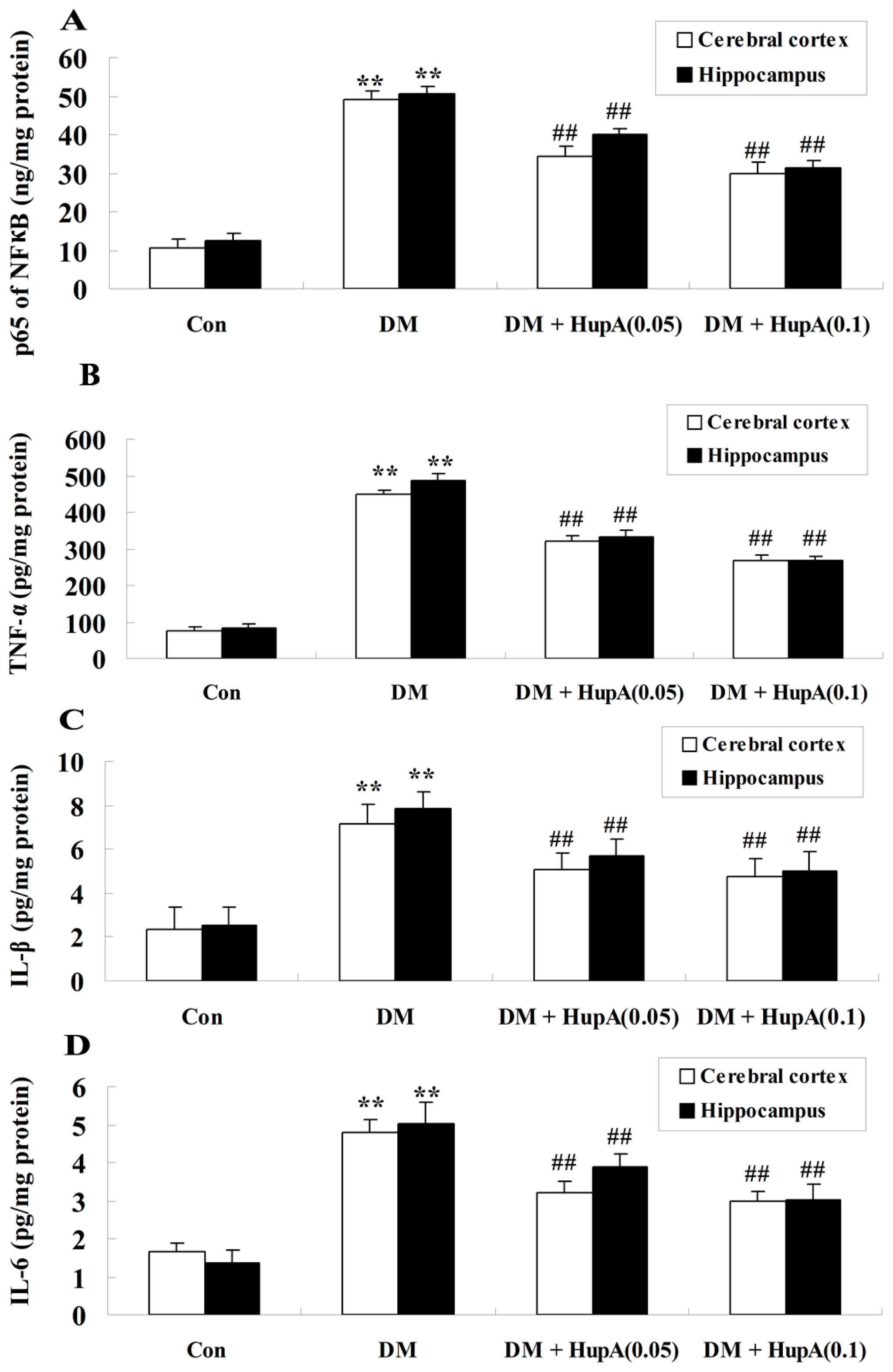
2.7. Effects of HupA on Inflammatory Cytokines in Diabetic Rat Brain
2.8. Effects of HupA on Caspase-3 Activity in Diabetic Rat Brain
3. Discussion
4. Experimental Section
4.1. Animals
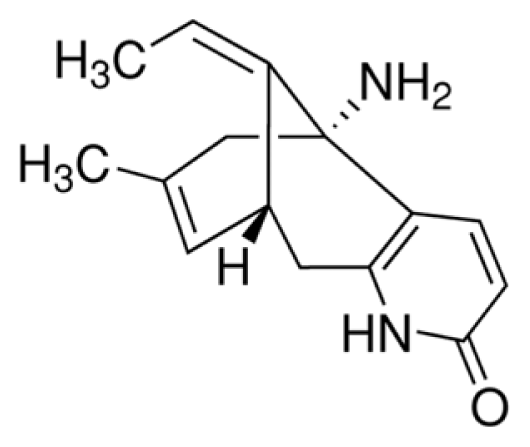
4.2. Chemicals and Drugs
4.3. Induction and Assessment of Diabetes
4.4. Morris Water Maze Test
4.5. Detection of Neuronal Loss in Hippocampus by Nissl Staining
4.6. Measurements of Acetylcholinesterase (AChE) and Choline Acetylase (ChAT) Activities
4.7. RNA Extraction and Quantitative Real-Time RT-PCR Analysis
4.8. Determination of BDNF Content
4.9. Assessment of Oxidative Stress
4.10. Measurements of Inflammatory Cytokines
4.11. Measurements of Caspase-3 Activity
4.12. Statistics
5. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsZhao-Qian Liu and Hong-Hao Zhou contributed to the study design. Xiao-Yuan Mao, Dan-Feng Cao, Xi Li, Ji-Ye Yin, Zhi-Bin Wang, Ying Zhang and Chen-Xue Mao performed the research and conducted the data analysis. Xiao-Yuan Mao wrote the manuscript.
References
- Biessels, G.J.; Deary, I.J.; Ryan, C.M. Cognition and diabetes: A lifespan perspective. Lancet Neurol 2008, 7, 184–190. [Google Scholar]
- Wrighten, S.A.; Piroli, G.G.; Grillo, C.A.; Reagan, L.P. A look inside the diabetic brain: Contributors to diabetes-induced brain aging. Biochim. Biophys. Acta 2009, 1792, 444–453. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C. Mechanistic insights into diabetes mellitus and oxidative stress. Curr. Med. Chem 2007, 14, 1729–1738. [Google Scholar]
- Mijnhout, G.S.; Scheltens, P.; Diamant, M.; Biessels, G.J.; Wessels, A.M.; Simsek, S.; Snoek, F.J.; Heine, R.J. Diabetic encephalopathy: A concept in need of a definition. Diabetologia 2006, 49, 1447–1448. [Google Scholar]
- Kodl, C.T.; Seaquist, E.R. Cognitive dysfunction and diabetes mellitus. Endocr. Rev 2008, 29, 494–511. [Google Scholar]
- Wessels, A.M.; Scheltens, P.; Barkhof, F.; Heine, R.J. Hyperglycaemia as a determinant of cognitive decline in patients with type 1 diabetes. Eur. J. Pharmacol 2008, 585, 88–96. [Google Scholar]
- Ryan, C.M.; Geckle, M.O.; Orchard, T.J. Cognitive efficiency declines over time in adults with Type 1 diabetes: Effects of micro- and macrovascular complications. Diabetologia 2003, 46, 940–948. [Google Scholar]
- Leibrock, J.; Lottspeich, F.; Hohn, A.; Hofer, M.; Hengerer, B.; Masiakowski, P.; Thoenen, H.; Barde, Y.A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature 1989, 341, 149–152. [Google Scholar]
- Xu, B.; Goulding, E.H.; Zang, K.; Cepoi, D.; Cone, R.D.; Jones, K.R.; Tecott, L.H.; Reichardt, L.F. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci 2003, 6, 736–742. [Google Scholar]
- Ono, M.; Ichihara, J.; Nonomura, T.; Itakura, Y.; Taiji, M.; Nakayama, C.; Noguchi, H. Brain-derived neurotrophic factor reduces blood glucose level in obese diabetic mice but not in normal mice. Biochem. Biophys. Res. Commun 1997, 238, 633–637. [Google Scholar]
- Liu, J.; Feng, L.; Ma, D.; Zhang, M.; Gu, J.; Wang, S.; Fu, Q.; Song, Y.; Lan, Z.; Qu, R.; et al. Neuroprotective effect of paeonol on cognition deficits of diabetic encephalopathy in streptozotocin-induced diabetic rat. Neurosci. Lett 2013, 549, 63–68. [Google Scholar]
- Mohamed, A.K.; Bierhaus, A.; Schiekofer, S.; Tritschler, H.; Ziegler, R.; Nawroth, P.P. The role of oxidative stress and NF-kappaB activation in late diabetic complications. Biofactors 1999, 10, 157–167. [Google Scholar]
- Liu, Y.W.; Zhu, X.; Li, W.; Lu, Q.; Wang, J.Y.; Wei, Y.Q.; Yin, X.X. Ginsenoside Re attenuates diabetes-associated cognitive deficits in rats. Pharmacol. Biochem. Behav 2012, 101, 93–98. [Google Scholar]
- Fukui, K.; Onodera, K.; Shinkai, T.; Suzuki, S.; Urano, S. Impairment of learning and memory in rats caused by oxidative stress and aging, and changes in antioxidative defense systems. Ann. N. Y. Acad. Sci 2001, 928, 168–175. [Google Scholar]
- Baydas, G.; Donder, E.; Kiliboz, M.; Sonkaya, E.; Tuzcu, M.; Yasar, A.; Nedzvetskii, V.S. Neuroprotection by alpha-lipoic acid in streptozotocin-induced diabetes. Biochemistry 2004, 69, 1001–1005. [Google Scholar]
- Deng, W.; Lu, H.; Teng, J. Carvacrol attenuates diabetes-associated cognitive deficits in rats. J. Mol. Neurosci 2013, 51, 813–819. [Google Scholar]
- Kuhad, A.; Bishnoi, M.; Tiwari, V.; Chopra, K. Suppression of NF-kappabeta signaling pathway by tocotrienol can prevent diabetes associated cognitive deficits. Pharmacol. Biochem. Behav 2009, 92, 251–259. [Google Scholar]
- Zhou, J.; Zhang, H.Y.; Tang, X.C. Huperzine A attenuates cognitive deficits and hippocampal neuronal damage after transient global ischemia in gerbils. Neurosci. Lett 2001, 313, 137–140. [Google Scholar]
- Wang, R.; Yan, H.; Tang, X.C. Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacol. Sin 2006, 27, 1–26. [Google Scholar]
- Wang, Z.F.; Tang, L.L.; Yan, H.; Wang, Y.J.; Tang, X.C. Effects of huperzine A on memory deficits and neurotrophic factors production after transient cerebral ischemia and reperfusion in mice. Pharmacol. Biochem. Behav 2006, 83, 603–611. [Google Scholar]
- Tang, L.L.; Wang, R.; Tang, X.C. Huperzine A protects SHSY5Y neuroblastoma cells against oxidative stress damage via nerve growth factor production. Eur. J. Pharmacol 2005, 519, 9–15. [Google Scholar]
- Wang, C.Y.; Zheng, W.; Wang, T.; Xie, J.W.; Wang, S.L.; Zhao, B.L.; Teng, W.P.; Wang, Z.Y. Huperzine A activates Wnt/beta-catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 2011, 36, 1073–1089. [Google Scholar]
- Forlenza, O.V.; Diniz, B.S.; Teixeira, A.L.; Ojopi, E.B.; Talib, L.L.; Mendonca, V.A.; Izzo, G.; Gattaz, W.F. Effect of brain-derived neurotrophic factor Val66Met polymorphism and serum levels on the progression of mild cognitive impairment. World J. Biol. Psychiatry 2010, 11, 774–780. [Google Scholar]
- Wu, W.; Wang, X.; Xiang, Q.; Meng, X.; Peng, Y.; Du, N.; Liu, Z.; Sun, Q.; Wang, C.; Liu, X. Astaxanthin alleviates brain aging in rats by attenuating oxidative stress and increasing BDNF levels. Food Funct 2013, 5, 158–166. [Google Scholar]
- Zhong, S.Z.; Ge, Q.H.; Qu, R.; Li, Q.; Ma, S.P. Paeonol attenuates neurotoxicity and ameliorates cognitive impairment induced by d-galactose in ICR mice. J. Neurol. Sci 2009, 277, 58–64. [Google Scholar]
- Cumiskey, D.; Butler, M.P.; Moynagh, P.N.; O’Connor, J. Evidence for a role for the group I metabotropic glutamate receptor in the inhibitory effect of tumor necrosis factor-alpha on long-term potentiation. Brain Res 2007, 1136, 13–19. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev 2004, 18, 2195–2224. [Google Scholar]
- Kim, E.K.; Kwon, K.B.; Han, M.J.; Song, M.Y.; Lee, J.H.; Lv, N.; Ka, S.O.; Yeom, S.R.; Kwon, Y.D.; Ryu, D.G.; et al. Coptidis rhizoma extract protects against cytokine-induced death of pancreatic beta-cells through suppression of NF-kappaB activation. Exp. Mol. Med 2007, 39, 149–159. [Google Scholar]
- Wang, J.; Zhang, H.Y.; Tang, X.C. Huperzine a improves chronic inflammation and cognitive decline in rats with cerebral hypoperfusion. J. Neurosci. Res 2010, 88, 807–815. [Google Scholar]
- Perry, G.; Castellani, R.J.; Hirai, K.; Smith, M.A. Reactive oxygen species mediate cellular damage in Alzheimer disease. J. Alzheimers Dis 1998, 1, 45–55. [Google Scholar]
- Shi, Q.; Fu, J.; Ge, D.; He, Y.; Ran, J.; Liu, Z.; Wei, J.; Diao, T.; Lu, Y. Huperzine A ameliorates cognitive deficits and oxidative stress in the hippocampus of rats exposed to acute hypobaric hypoxia. Neurochem. Res 2012, 37, 2042–2052. [Google Scholar]
- Zhou, J.; Tang, X.C. Huperzine A attenuates apoptosis and mitochondria-dependent caspase-3 in rat cortical neurons. FEBS Lett 2002, 526, 21–25. [Google Scholar]
- Kuhad, A.; Chopra, K. Effect of sesamol on diabetes-associated cognitive decline in rats. Exp. Brain Res 2008, 185, 411–420. [Google Scholar]
- Morris, R.G.; Garrud, P.; Rawlins, J.N.; O’Keefe, J. Place navigation impaired in rats with hippocampal lesions. Nature 1982, 297, 681–683. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Body weight (g) | Plasma glucose (mg/dL) | ||
|---|---|---|---|---|
| Onset of study | End of study | Onset of study | End of study | |
| Con | 242.20 ± 4.38 | 285.70 ± 4.27 | 114.30 ± 2.57 | 108.00 ± 1.53 |
| DM | 245.23 ± 5.31 | 140.21 ± 4.89 ** | 110.14 ± 2.62 | 585.10 ± 3.68 ** |
| DM + Hup (0.05) | 239.90 ± 5.36 | 232.50 ± 3.28 ## | 106.25 ± 2.79 | 306.90 ± 3.87 ## |
| DM + HupA (0.1) | 240.50 ± 5.43 | 256.40 ± 5.25 ## | 107.48 ± 2.39 | 299.00 ± 3.48 ## |
| Gene | Primer sequences(5′-to-3′) | PCR product size | Accession number |
|---|---|---|---|
| BDNF | Forward:5′- ATGGGTTACACGAAGGAAGG -3′ Reverse:5′- CCGAACATACGATTGGGTAGT -3′ | 84 bp | NM_012513.3 |
| β-actin | Forward:5′- AGGCCCCTCTGAACCCTAAG -3′ Reverse:5′- CCAGAGGCATACAGGGACAAC -3′ | 118 bp | EF156276 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mao, X.-Y.; Cao, D.-F.; Li, X.; Yin, J.-Y.; Wang, Z.-B.; Zhang, Y.; Mao, C.-X.; Zhou, H.-H.; Liu, Z.-Q. Huperzine A Ameliorates Cognitive Deficits in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2014, 15, 7667-7683. https://doi.org/10.3390/ijms15057667
Mao X-Y, Cao D-F, Li X, Yin J-Y, Wang Z-B, Zhang Y, Mao C-X, Zhou H-H, Liu Z-Q. Huperzine A Ameliorates Cognitive Deficits in Streptozotocin-Induced Diabetic Rats. International Journal of Molecular Sciences. 2014; 15(5):7667-7683. https://doi.org/10.3390/ijms15057667
Chicago/Turabian StyleMao, Xiao-Yuan, Dan-Feng Cao, Xi Li, Ji-Ye Yin, Zhi-Bin Wang, Ying Zhang, Chen-Xue Mao, Hong-Hao Zhou, and Zhao-Qian Liu. 2014. "Huperzine A Ameliorates Cognitive Deficits in Streptozotocin-Induced Diabetic Rats" International Journal of Molecular Sciences 15, no. 5: 7667-7683. https://doi.org/10.3390/ijms15057667