Marine Microbial Metagenomics: From Individual to the Environment


{kind=link}
Abstract
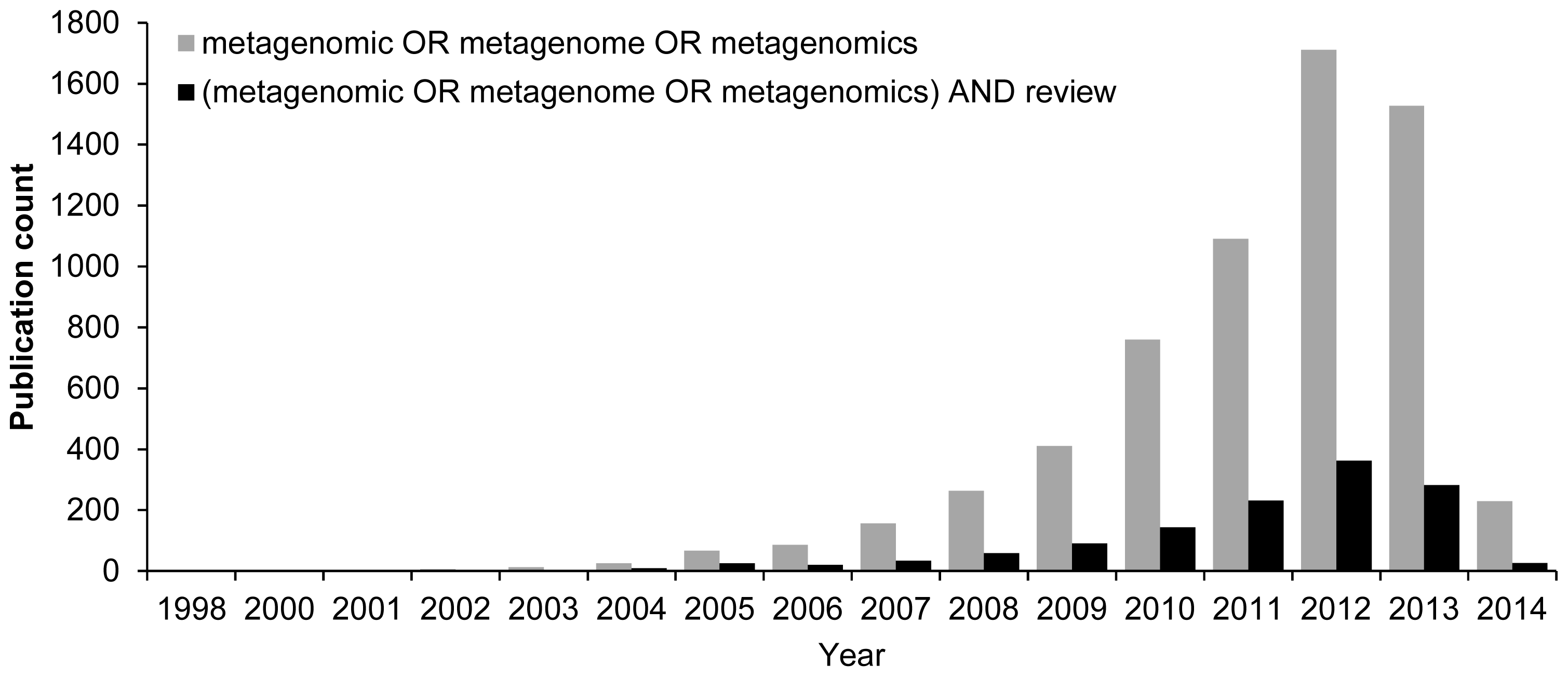
:1. Introduction
2. Environmental Ecology Derived from Functional Genomics
3. Microbial Plasmid Metagenomics
4. Metabolism and Element Cycling Discovered in Metagenomics Studies
5. Investigation of Microbial Communities Using Comparative Metagenomics
6. Decoupling of Community and Functional Diversity
7. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsC.-H.T. and S.-L.T. draft, read, and approved the manuscript.
References
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol 1998, 5, 245–249. [Google Scholar]
- Hugenholtz, P.; Goebel, B.M.; Pace, N.R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol 1998, 180, 4765–4774. [Google Scholar]
- Stein, J.L.; Marsh, T.L.; Wu, K.Y.; Shizuya, H.; DeLong, E.F. Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J. Bacteriol 1996, 178, 591–599. [Google Scholar]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar]
- PubMed. Availble online: http://www.ncbi.nlm.nih.gov/pubmed/ accessed on 14 March 2014.
- DeLong, E.F. Microbial community genomics in the ocean. Nat. Rev. Microbiol 2005, 3, 459–469. [Google Scholar]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev 2004, 68, 669–685. [Google Scholar]
- Singh, J.; Behal, A.; Singla, N.; Joshi, A.; Birbian, N.; Singh, S.; Bali, V.; Batra, N. Metagenomics: Concept, methodology, ecological inference and recent advances. Biotechnol. J 2009, 4, 480–494. [Google Scholar]
- Hugenholtz, P.; Tyson, G.W. Microbiology: Metagenomics. Nature 2008, 455, 481–483. [Google Scholar]
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. Metagenomics: Genomic analysis of microbial communities. Annu. Rev. Genet 2004, 38, 525–552. [Google Scholar]
- Schloss, P.D.; Handelsman, J. Metagenomics for studying unculturable microorganisms: Cutting the Gordian knot. Genome Biol 2005, 6. [Google Scholar] [CrossRef]
- Wooley, J.C.; Godzik, A.; Friedberg, I. A primer on metagenomics. PLoS Comp. Biol 2010, 6, e1000667. [Google Scholar]
- Gilbert, J.A.; Dupont, C.L. Microbial metagenomics: Beyond the genome. Annu. Rev. Mar. Sci 2011, 3, 347–371. [Google Scholar]
- Xu, J. Microbial ecology in the age of genomics and metagenomics: Concepts, tools, and recent advances. Mol. Ecol 2006, 15, 1713–1731. [Google Scholar]
- Lindell, D.; Jaffe, J.D.; Johnson, Z.I.; Church, G.M.; Chisholm, S.W. Photosynthesis genes in marine viruses yield proteins during host infection. Nature 2005, 438, 86–89. [Google Scholar]
- Sullivan, M.B.; Lindell, D.; Lee, J.A.; Thompson, L.R.; Bielawski, J.P.; Chisholm, S.W. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol 2006, 4, e234. [Google Scholar]
- Chenard, C.; Suttle, C.A. Phylogenetic diversity of sequences of cyanophage photosynthetic gene psbA in marine and freshwaters. Appl. Environ. Microbiol 2008, 74, 5317–5324. [Google Scholar]
- Tripp, H.J.; Bench, S.R.; Turk, K.A.; Foster, R.A.; Desany, B.A.; Niazi, F.; Affourtit, J.P.; Zehr, J.P. Metabolic streamlining in an open-ocean nitrogen-fixing cyanobacterium. Nature 2010, 464, 90–94. [Google Scholar]
- Zhang, S.; Bryant, D.A. The tricarboxylic acid cycle in cyanobacteria. Science 2011, 334, 1551–1553. [Google Scholar]
- Dufresne, A.; Salanoubat, M.; Partensky, F.; Artiguenave, F.; Axmann, I.M.; Barbe, V.; Duprat, S.; Galperin, M.Y.; Koonin, E.V.; le Gall, F.; et al. Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc. Natl. Acad. Sci. USA 2003, 100, 10020–10025. [Google Scholar]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar]
- Coleman, M.L.; Sullivan, M.B.; Martiny, A.C.; Steglich, C.; Barry, K.; Delong, E.F.; Chisholm, S.W. Genomic islands and the ecology and evolution of Prochlorococcus. Science 2006, 311, 1768–1770. [Google Scholar]
- Giovannoni, S.J.; Tripp, H.J.; Givan, S.; Podar, M.; Vergin, K.L.; Baptista, D.; Bibbs, L.; Eads, J.; Richardson, T.H.; Noordewier, M.; et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science 2005, 309, 1242–1245. [Google Scholar]
- Moran, M.A.; Buchan, A.; Gonzalez, J.M.; Heidelberg, J.F.; Whitman, W.B.; Kiene, R.P.; Henriksen, J.R.; King, G.M.; Belas, R.; Fuqua, C.; et al. Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 2004, 432, 910–913. [Google Scholar]
- Woyke, T.; Xie, G.; Copeland, A.; Gonzalez, J.M.; Han, C.; Kiss, H.; Saw, J.H.; Senin, P.; Yang, C.; Chatterji, S.; et al. Assembling the marine metagenome, one cell at a time. PLoS One 2009, 4, e5299. [Google Scholar]
- Yooseph, S.; Sutton, G.; Rusch, D.B.; Halpern, A.L.; Williamson, S.J.; Remington, K.; Eisen, J.A.; Heidelberg, K.B.; Manning, G.; Li, W.; et al. The Sorcerer II Global Ocean Sampling expedition: Expanding the universe of protein families. PLoS Biol 2007, 5, e16. [Google Scholar]
- Rusch, D.B.; Halpern, A.L.; Sutton, G.; Heidelberg, K.B.; Williamson, S.; Yooseph, S.; Wu, D.; Eisen, J.A.; Hoffman, J.M.; Remington, K.; et al. The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol 2007, 5, e77. [Google Scholar]
- Rusch, D.B.; Martiny, A.C.; Dupont, C.L.; Halpern, A.L.; Venter, J.C. Characterization of Prochlorococcus clades from iron-depleted oceanic regions. Proc. Natl. Acad. Sci. USA 2010, 107, 16184–16189. [Google Scholar]
- Eloe, E.A.; Lauro, F.M.; Vogel, R.F.; Bartlett, D.H. The deep-sea bacterium Photobacterium profundum SS9 utilizes separate flagellar systems for swimming and swarming under high-pressure conditions. Appl. Environ. Microbiol 2008, 74, 6298–6305. [Google Scholar]
- Vezzi, A.; Campanaro, S.; D’Angelo, M.; Simonato, F.; Vitulo, N.; Lauro, F.M.; Cestaro, A.; Malacrida, G.; Simionati, B.; Cannata, N.; et al. Life at depth: Photobacterium profundum genome sequence and expression analysis. Science 2005, 307, 1459–1461. [Google Scholar]
- Methe, B.A.; Nelson, K.E.; Deming, J.W.; Momen, B.; Melamud, E.; Zhang, X.J.; Moult, J.; Madupu, R.; Nelson, W.C.; Dodson, R.J.; et al. The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proc. Natl. Acad. Sci. USA 2005, 102, 10913–10918. [Google Scholar]
- Medigue, C.; Krin, E.; Pascal, G.; Barbe, V.; Bernsel, A.; Bertin, P.N.; Cheung, F.; Cruveiller, S.; D’Amico, S.; Duilio, A.; et al. Coping with cold: The genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res 2005, 15, 1325–1335. [Google Scholar]
- Cook, M.A.; Osborn, A.M.; Bettandorff, J.; Sobecky, P.A. Endogenous isolation of replicon probes for assessing plasmid ecology of marine sediment microbial communities. Microbiology 2001, 147, 2089–2101. [Google Scholar]
- Moran, M.A.; Belas, R.; Schell, M.A.; Gonzalez, J.M.; Sun, F.; Sun, S.; Binder, B.J.; Edmonds, J.; Ye, W.; Orcutt, B.; et al. Ecological genomics of marine Roseobacters. Appl. Environ. Microbiol 2007, 73, 4559–4569. [Google Scholar]
- Szczepanowski, R.; Bekel, T.; Goesmann, A.; Krause, L.; Kromeke, H.; Kaiser, O.; Eichler, W.; Puhler, A.; Schluter, A. Insight into the plasmid metagenome of wastewater treatment plant bacteria showing reduced susceptibility to antimicrobial drugs analysed by the 454-pyrosequencing technology. J. Biotechnol 2008, 136, 54–64. [Google Scholar]
- Zhang, T.; Zhang, X.X.; Ye, L. Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge. PLoS One 2011, 6, e26041. [Google Scholar]
- Sentchilo, V.; Mayer, A.P.; Guy, L.; Miyazaki, R.; Tringe, S.G.; Barry, K.; Malfatti, S.; Goessmann, A.; Robinson-Rechavi, M.; van der Meer, J.R. Community-wide plasmid gene mobilization and selection. ISME J 2013, 7, 1173–1186. [Google Scholar]
- Kav, A.B.; Sasson, G.; Jami, E.; Doron-Faigenboim, A.; Benhar, I.; Mizrahi, I. Insights into the bovine rumen plasmidome. Proc. Natl. Acad. Sci. USA 2012, 109, 5452–5457. [Google Scholar]
- Beja, O.; Aravind, L.; Koonin, E.V.; Suzuki, M.T.; Hadd, A.; Nguyen, L.P.; Jovanovich, S.B.; Gates, C.M.; Feldman, R.A.; Spudich, J.L.; et al. Bacterial rhodopsin: Evidence for a new type of phototrophy in the sea. Science 2000, 289, 1902–1906. [Google Scholar]
- Man, D.; Wang, W.; Sabehi, G.; Aravind, L.; Post, A.F.; Massana, R.; Spudich, E.N.; Spudich, J.L.; Beja, O. Diversification and spectral tuning in marine proteorhodopsins. EMBO J 2003, 22, 1725–1731. [Google Scholar]
- Schleper, C.; Jurgens, G.; Jonuscheit, M. Genomic studies of uncultivated archaea. Nat. Rev. Microbiol 2005, 3, 479–488. [Google Scholar]
- Konneke, M.; Bernhard, A.E.; de la Torre, J.R.; Walker, C.B.; Waterbury, J.B.; Stahl, D.A. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 2005, 437, 543–546. [Google Scholar]
- Hallam, S.J.; Mincer, T.J.; Schleper, C.; Preston, C.M.; Roberts, K.; Richardson, P.M.; DeLong, E.F. Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota. PLoS Biol 2006, 4, e95. [Google Scholar]
- Sebastian, M.; Ammerman, J.W. The alkaline phosphatase PhoX is more widely distributed in marine bacteria than the classical PhoA. ISME J 2009, 3, 563–572. [Google Scholar]
- Luo, H.; Benner, R.; Long, R.A.; Hu, J. Subcellular localization of marine bacterial alkaline phosphatases. Proc. Natl. Acad. Sci. USA 2009, 106, 21219–21223. [Google Scholar]
- Martiny, A.C.; Huang, Y.; Li, W. Occurrence of phosphate acquisition genes in Prochlorococcus cells from different ocean regions. Environ. Microbiol 2009, 11, 1340–1347. [Google Scholar]
- Gilbert, J.A.; Thomas, S.; Cooley, N.A.; Kulakova, A.; Field, D.; Booth, T.; McGrath, J.W.; Quinn, J.P.; Joint, I. Potential for phosphonoacetate utilization by marine bacteria in temperate coastal waters. Environ. Microbiol 2009, 11, 111–125. [Google Scholar]
- Martinez, A.; Tyson, G.W.; Delong, E.F. Widespread known and novel phosphonate utilization pathways in marine bacteria revealed by functional screening and metagenomic analyses. Environ. Microbiol 2010, 12, 222–238. [Google Scholar] [Green Version]
- Yu, X.; Doroghazi, J.R.; Janga, S.C.; Zhang, J.K.; Circello, B.; Griffin, B.M.; Labeda, D.P.; Metcalf, W.W. Diversity and abundance of phosphonate biosynthetic genes in nature. Proc. Natl. Acad. Sci. USA 2013, 110, 20759–20764. [Google Scholar]
- Villarreal-Chiu, J.F.; Quinn, J.P.; McGrath, J.W. The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front. Microbiol 2012, 3. [Google Scholar] [CrossRef]
- Feingersch, R.; Philosof, A.; Mejuch, T.; Glaser, F.; Alalouf, O.; Shoham, Y.; Beja, O. Potential for phosphite and phosphonate utilization by Prochlorococcus. ISME J 2012, 6, 827–834. [Google Scholar]
- Howard, E.C.; Sun, S.; Biers, E.J.; Moran, M.A. Abundant and diverse bacteria involved in DMSP degradation in marine surface waters. Environ. Microbiol 2008, 10, 2397–2410. [Google Scholar]
- Todd, J.D.; Curson, A.R.; Dupont, C.L.; Nicholson, P.; Johnston, A.W. The dddP gene, encoding a novel enzyme that converts dimethylsulfoniopropionate into dimethyl sulfide, is widespread in ocean metagenomes and marine bacteria and also occurs in some Ascomycete fungi. Environ. Microbiol 2009, 11, 1376–1385. [Google Scholar]
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.E.; Ram, R.J.; Richardson, P.M.; Solovyev, V.V.; Rubin, E.M.; Rokhsar, D.S.; Banfield, J.F. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37–43. [Google Scholar]
- Tringe, S.G.; von Mering, C.; Kobayashi, A.; Salamov, A.A.; Chen, K.; Chang, H.W.; Podar, M.; Short, J.M.; Mathur, E.J.; Detter, J.C.; et al. Comparative metagenomics of microbial communities. Science 2005, 308, 554–557. [Google Scholar]
- Foerstner, K.U.; von Mering, C.; Hooper, S.D.; Bork, P. Environments shape the nucleotide composition of genomes. EMBO Rep 2005, 6, 1208–1213. [Google Scholar]
- DeLong, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Frigaard, N.U.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar]
- Dinsdale, E.A.; Edwards, R.A.; Hall, D.; Angly, F.; Breitbart, M.; Brulc, J.M.; Furlan, M.; Desnues, C.; Haynes, M.; Li, L.; et al. Functional metagenomic profiling of nine biomes. Nature 2008, 452, 629–632. [Google Scholar]
- Willner, D.; Thurber, R.V.; Rohwer, F. Metagenomic signatures of 86 microbial and viral metagenomes. Environ. Microbiol 2009, 11, 1752–1766. [Google Scholar]
- Lauro, F.M.; McDougald, D.; Thomas, T.; Williams, T.J.; Egan, S.; Rice, S.; DeMaere, M.Z.; Ting, L.; Ertan, H.; Johnson, J.; et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 15527–15533. [Google Scholar]
- Yooseph, S.; Nealson, K.H.; Rusch, D.B.; McCrow, J.P.; Dupont, C.L.; Kim, M.; Johnson, J.; Montgomery, R.; Ferriera, S.; Beeson, K.; et al. Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature 2010, 468, 60–66. [Google Scholar]
- Swan, B.K.; Tupper, B.; Sczyrba, A.; Lauro, F.M.; Martinez-Garcia, M.; Gonzalez, J.M.; Luo, H.; Wright, J.J.; Landry, Z.C.; Hanson, N.W.; et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc. Natl. Acad. Sci. USA 2013, 110, 11463–11468. [Google Scholar] [Green Version]
- Brazelton, W.J.; Baross, J.A. Abundant transposases encoded by the metagenome of a hydrothermal chimney biofilm. ISME J 2009, 3, 1420–1424. [Google Scholar]
- Konstantinidis, K.T.; Braff, J.; Karl, D.M.; DeLong, E.F. Comparative metagenomic analysis of a microbial community residing at a depth of 4,000 meters at station ALOHA in the North Pacific subtropical gyre. Appl. Environ. Microbiol 2009, 75, 5345–5355. [Google Scholar]
- Walsh, D.A.; Zaikova, E.; Howes, C.G.; Song, Y.C.; Wright, J.J.; Tringe, S.G.; Tortell, P.D.; Hallam, S.J. Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones. Science 2009, 326, 578–582. [Google Scholar]
- Ganesh, S.; Parris, D.J.; DeLong, E.F.; Stewart, F.J. Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone. ISME J 2014, 8, 187–211. [Google Scholar]
- Frias-Lopez, J.; Shi, Y.; Tyson, G.W.; Coleman, M.L.; Schuster, S.C.; Chisholm, S.W.; DeLong, E.F. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. USA 2008, 105, 3805–3810. [Google Scholar]
- Hewson, I.; Poretsky, R.S.; Tripp, H.J.; Montoya, J.P.; Zehr, J.P. Spatial patterns and light-driven variation of microbial population gene expression in surface waters of the oligotrophic open ocean. Environ. Microbiol 2010, 12, 1940–1956. [Google Scholar]
- Bell, T.; Newman, J.A.; Silverman, B.W.; Turner, S.L.; Lilley, A.K. The contribution of species richness and composition to bacterial services. Nature 2005, 436, 1157–1160. [Google Scholar]
- Hewson, I.; Paerl, R.W.; Tripp, H.J.; Zehr, J.P.; Karl, D.M. Metagenomic potential of microbial assemblages in the surface waters of the central Pacific Ocean tracks variability in oceanic habitat. Limnol. Oceanogr 2009, 54, 1981–1994. [Google Scholar]
- Burke, C.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. USA 2011, 108, 14288–14293. [Google Scholar]
- Tseng, C.H.; Chiang, P.W.; Shiah, F.K.; Chen, Y.L.; Liou, J.R.; Hsu, T.C.; Maheswararajah, S.; Saeed, I.; Halgamuge, S.; Tang, S.L. Microbial and viral metagenomes of a subtropical freshwater reservoir subject to climatic disturbances. ISME J 2013, 7, 2374–2386. [Google Scholar]
- Frossard, A.; Gerull, L.; Mutz, M.; Gessner, M.O. Disconnect of microbial structure and function: Enzyme activities and bacterial communities in nascent stream corridors. ISME J 2012, 6, 680–691. [Google Scholar]
- Allison, S.D.; Martiny, J.B.H. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar]

© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tseng, C.-H.; Tang, S.-L. Marine Microbial Metagenomics: From Individual to the Environment. Int. J. Mol. Sci. 2014, 15, 8878-8892. https://doi.org/10.3390/ijms15058878
Tseng C-H, Tang S-L. Marine Microbial Metagenomics: From Individual to the Environment. International Journal of Molecular Sciences. 2014; 15(5):8878-8892. https://doi.org/10.3390/ijms15058878
Chicago/Turabian StyleTseng, Ching-Hung, and Sen-Lin Tang. 2014. "Marine Microbial Metagenomics: From Individual to the Environment" International Journal of Molecular Sciences 15, no. 5: 8878-8892. https://doi.org/10.3390/ijms15058878
APA StyleTseng, C.-H., & Tang, S.-L. (2014). Marine Microbial Metagenomics: From Individual to the Environment. International Journal of Molecular Sciences, 15(5), 8878-8892. https://doi.org/10.3390/ijms15058878