Brain Metastasis-Initiating Cells: Survival of the Fittest

Abstract
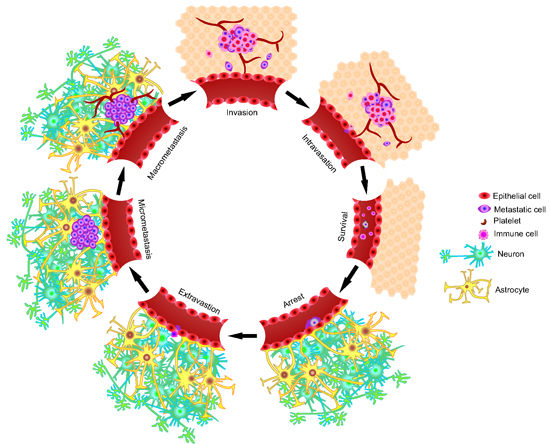
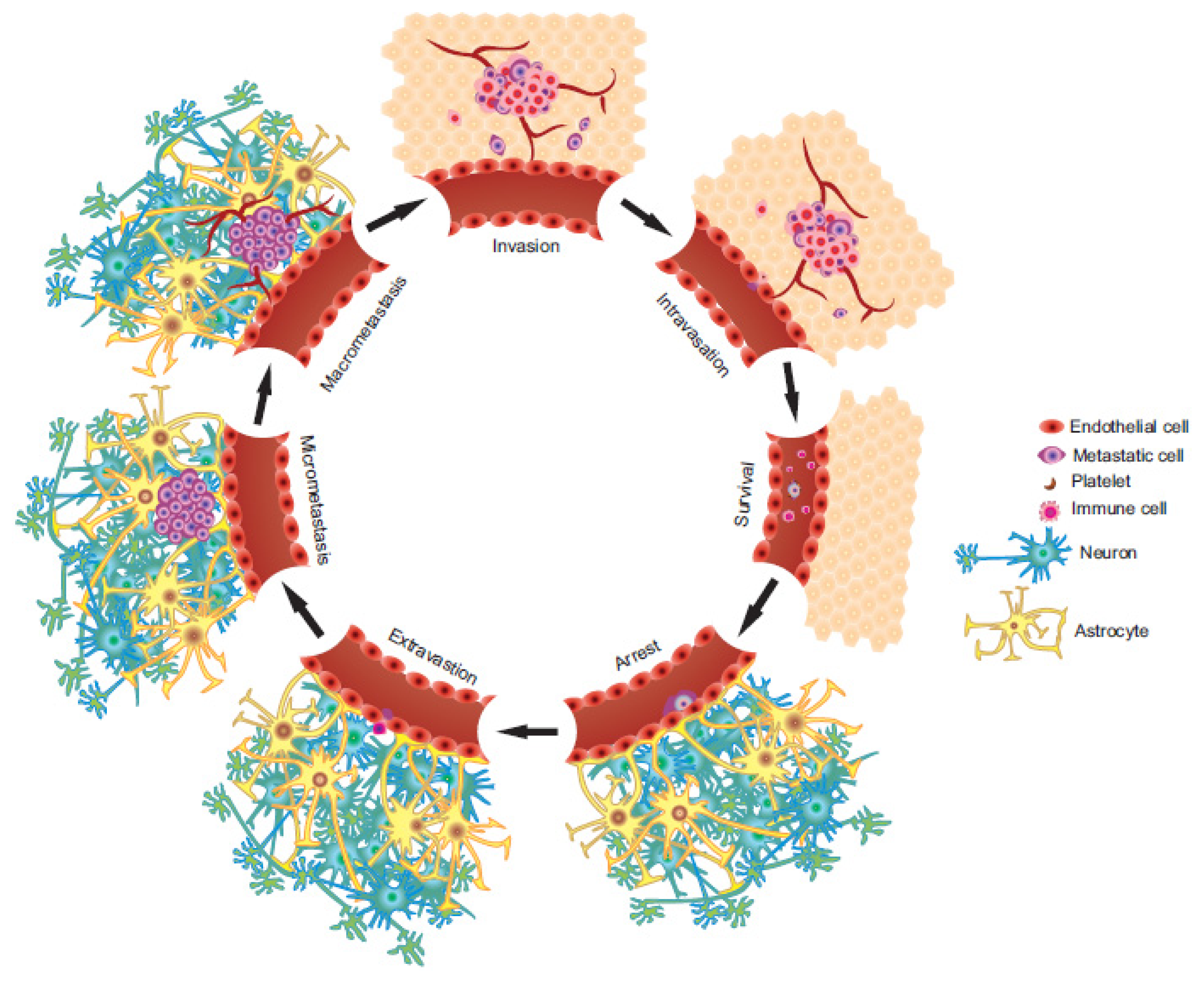
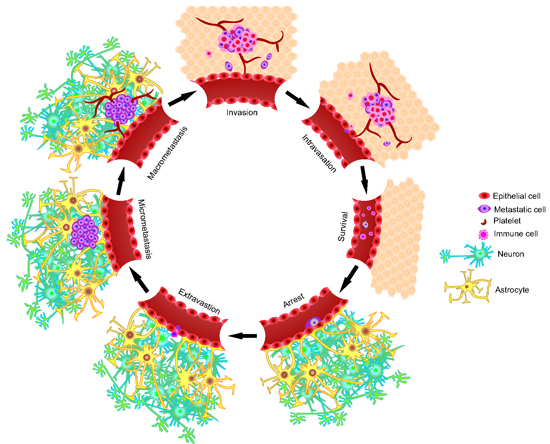
:
1. Introduction
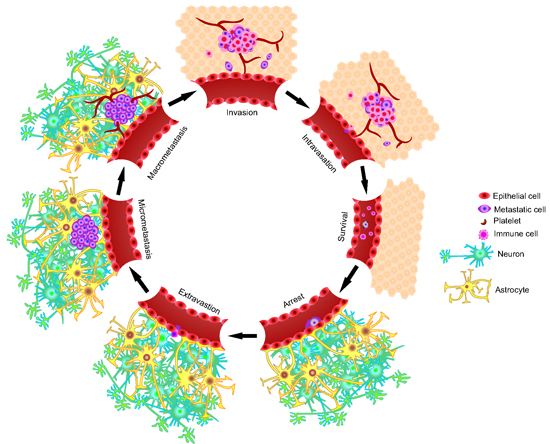
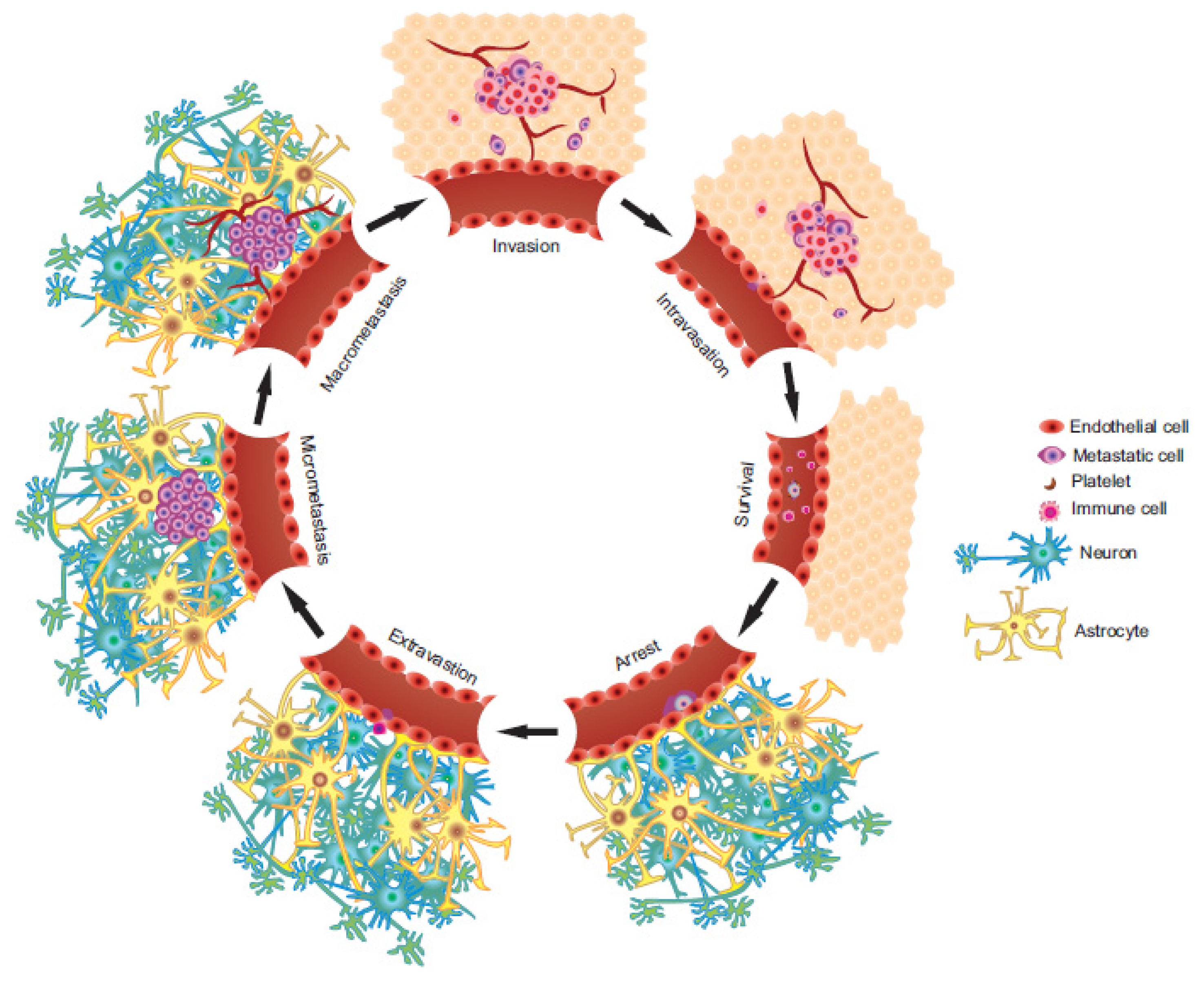
2. The Metastatic Process
2.1. Invasion/Migration
2.2. Intravasation
2.3. Circulation
2.4. Arrest and Extravasation
2.5. Colonization of the Secondary Microenvironment
3. Frameworks for Studying Metastasis
3.1. Seed/Soil Hypothesis
3.2. Mechanical Hypothesis
3.3. Epithelial–Mesenchymal Transition
3.4. Cancer Stem Cell Hypothesis
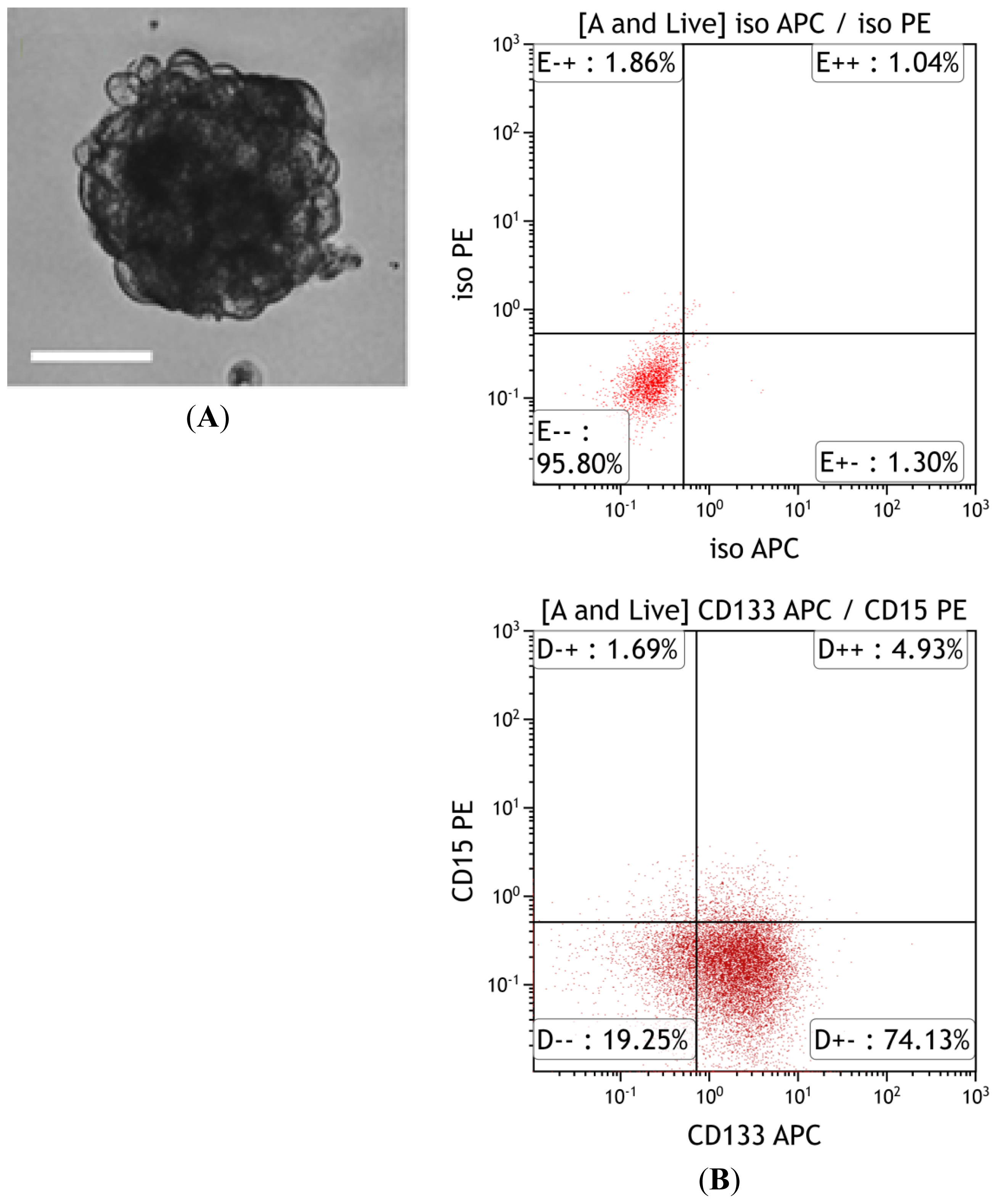
3.5. In Vivo Validation of Tumor-Initiating Capacity
3.6. Potential Therapeutic Targets in Brain Metastasis-Initiating Cells
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsM.S., B.M., and S.K.S. co-conceived the review. M.S. and B.M. wrote the review. S.M, N.M, and C.V. critically revised the manuscript. M.S., N.M., and C.V. developed the figures for the manuscript. All authors approved the manuscript for submission.
References
- Langley, R.R.; Fidler, I.J. The biology of brain metastasis. Clin. Chem 2013, 59, 180–189. [Google Scholar]
- Rahmathulla, G.; Toms, S.A.; Weil, R.J. The molecular biology of brain metastasis. J. Oncol 2012, 2012, 723541. [Google Scholar]
- Sjobakk, T.E.; Vettukattil, R.; Gulati, M.; Gulati, S.; Lundgren, S.; Gribbestad, I.S.; Torp, S.H.; Bathen, T.F. Metabolic profiles of brain metastases. Int. J. Mol. Sci 2013, 14, 2104–2118. [Google Scholar]
- Kress, M.A.; Oermann, E.; Ewend, M.G.; Hoffman, R.B.; Chaudhry, H.; Collins, B. Stereotactic radiosurgery for single brain metastases from non-small cell lung cancer: Progression of extracranial disease correlates with distant intracranial failure. Radiat. Oncol 2013, 8, 64. [Google Scholar]
- Palmieri, D. An introduction to brain metastasis. In Central Nervous System Metastasis, the Biological Basis and Clinical Considerations; Palmieri, D., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 1–13. [Google Scholar]
- Patchell, R.A. The management of brain metastases. Cancer Treat. Rev 2003, 29, 533–540. [Google Scholar]
- Soffietti, R.; Ruda, R.; Mutani, R. Management of brain metastases. J. Neurol 2002, 249, 1357–1369. [Google Scholar]
- Delattre, J.Y.; Krol, G.; Thaler, H.T.; Posner, J.B. Distribution of brain metastases. Arch. Neurol 1988, 45, 741–744. [Google Scholar]
- Berghoff, A.S.; Rajky, O.; Winkler, F.; Bartsch, R.; Furtner, J.; Hainfellner, J.A.; Goodman, S.L.; Weller, M.; Schittenhelm, J.; Preusser, M. Invasion patterns in brain metastases of solid cancers. Neuro Oncol 2013, 15, 1664–1672. [Google Scholar]
- Weiss, L. Comments on hematogenous metastatic patterns in humans as revealed by autopsy. Clin. Exp. Metastasis 1992, 10, 191–199. [Google Scholar]
- Chiang, A.C.; Massague, J. Molecular basis of metastasis. N. Engl. J. Med 2008, 359, 2814–2823. [Google Scholar]
- Cameron, M.D.; Schmidt, E.E.; Kerkvliet, N.; Nadkarni, K.V.; Morris, V.L.; Groom, A.C.; Chambers, A.F.; MacDonald, I.C. Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 2000, 60, 2541–2546. [Google Scholar]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol 1998, 153, 865–873. [Google Scholar]
- Fidler, I.J. Cancer metastasis. Br. Med. Bull 1991, 47, 157–177. [Google Scholar]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med 2006, 12, 895–904. [Google Scholar]
- Beasley, K.D.; Toms, S.A. The molecular pathobiology of metastasis to the brain: A review. Neurosurg. Clin. N. Am 2011, 22, 7–14. [Google Scholar]
- Yoshimasu, T.; Sakurai, T.; Oura, S.; Hirai, I.; Tanino, H.; Kokawa, Y.; Naito, Y.; Okamura, Y.; Ota, I.; Tani, N.; et al. Increased expression of integrin α3β1 in highly brain metastatic subclone of a human non-small cell lung cancer cell line. Cancer Sci 2004, 95, 142–148. [Google Scholar]
- Gavrilovic, I.T.; Posner, J.B. Brain metastases: Epidemiology and pathophysiology. J. Neuro Oncol 2005, 75, 5–14. [Google Scholar]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med 2013, 19, 1423–1437. [Google Scholar]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Mannel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 1999, 59, 1295–1300. [Google Scholar]
- Grossmann, J. Molecular mechanisms of detachment-induced apoptosis—Anoikis. Apoptosis 2002, 7, 247–260. [Google Scholar]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med 2010, 16, 116–122. [Google Scholar]
- Ramakrishna, R.; Rostomily, R. Seed, soil, and beyond: The basic biology of brain metastasis. Surg. Neurol. Int 2013, 4, S256–S264. [Google Scholar]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar]
- Fayard, B.; Bianchi, F.; Dey, J.; Moreno, E.; Djaffer, S.; Hynes, N.E.; Monard, D. The serine protease inhibitor protease nexin-1 controls mammary cancer metastasis through LRP-1-mediated MMP-9 expression. Cancer Res 2009, 69, 5690–5698. [Google Scholar]
- Shellenberger, T.D.; Mazumdar, A.; Henderson, Y.; Briggs, K.; Wang, M.; Chattopadhyay, C.; Jayakumar, A.; Frederick, M.; Clayman, G.L. Headpin: A serpin with endogenous and exogenous suppression of angiogenesis. Cancer Res 2005, 65, 11501–11509. [Google Scholar]
- Yin, S.; Lockett, J.; Meng, Y.; Biliran, H., Jr.; Blouse, G.E.; Li, X.; Reddy, N.; Zhao, Z.; Lin, X.; Anagli, J.; et al. Maspin retards cell detachment via a novel interaction with the urokinase-type plasminogen activator/urokinase-type plasminogen activator receptor system. Cancer Res 2006, 66, 4173–4181. [Google Scholar]
- Xiao, G.; Liu, Y.E.; Gentz, R.; Sang, Q.A.; Ni, J.; Goldberg, I.D.; Shi, Y.E. Suppression of breast cancer growth and metastasis by a serpin myoepithelium-derived serine proteinase inhibitor expressed in the mammary myoepithelial cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3700–3705. [Google Scholar]
- Vanharanta, S.; Massague, J. Origins of metastatic traits. Cancer Cell 2013, 24, 410–421. [Google Scholar]
- Fidler, I.J.; Schackert, G.; Zhang, R.D.; Radinsky, R.; Fujimaki, T. The biology of melanoma brain metastasis. Cancer Metastasis Rev 1999, 18, 387–400. [Google Scholar]
- Stephen Paget’s paper reproduced from The Lancet, 1889. Cancer Metastasis Rev 1989, 8, 98–101.
- Fidler, I.J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar]
- James Ewing, A.M. Neoplastic diseases: A treatise on tumours. Br. J. Surg 1928, 16, 174–175. [Google Scholar]
- Weiss, L. Metastasis of cancer: A conceptual history from antiquity to the 1990s. Cancer Metastasis Rev 2000, 19, 193–383. [Google Scholar]
- Sugarbaker, E.V. Cancer metastasis: A product of tumor-host interactions. Curr. Probl. Cancer 1979, 3, 1–59. [Google Scholar]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar]
- Lillie, F.R. The Development of the Chick: An Introduction to Embryology, 2nd ed; Henry Holt: New York, NY, USA, 1936. [Google Scholar]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol 2012, 22, 396–403. [Google Scholar]
- Kong, D.; Li, Y.; Wang, Z.; Sarkar, F.H. Cancer stem cells and epithelial-to-mesenchymal transition (EMT)-phenotypic cells: Are they cousins or twins? Cancers 2011, 3, 716–729. [Google Scholar]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol 2005, 17, 548–558. [Google Scholar]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable β-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008, 3, e2888. [Google Scholar]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res 2006, 66, 9339–9344. [Google Scholar]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 1997, 3, 730–737. [Google Scholar]
- Ailles, L.E.; Weissman, I.L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol 2007, 18, 460–466. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005, 65, 10946–10951. [Google Scholar]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar]
- Goodison, S.; Kawai, K.; Hihara, J.; Jiang, P.; Yang, M.; Urquidi, V.; Hoffman, R.M.; Tarin, D. Prolonged dormancy and site-specific growth potential of cancer cells spontaneously disseminated from nonmetastatic breast tumors as revealed by labeling with green fluorescent protein. Clin. Cancer Res 2003, 9, 3808–3814. [Google Scholar]
- Celia-Terrassa, T.; Meca-Cortes, O.; Mateo, F.; de Paz, A.M.; Rubio, N.; Arnal-Estape, A.; Ell, B.J.; Bermudo, R.; Diaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig 2012, 122, 1849–1868. [Google Scholar]
- Nolte, S.M.; Venugopal, C.; McFarlane, N.; Morozova, O.; Hallett, R.M.; O’Farrell, E.; Manoranjan, B.; Murty, N.K.; Klurfan, P.; Kachur, E.; et al. A cancer stem cell model for studying brain metastases from primary lung cancer. J. Natl. Cancer Inst 2013, 105, 551–562. [Google Scholar]
- Todorutiu, C.; Risca, R. Experimental studies on factors influencing the widespread organ distribution of Walker 256 tumour metastases. Morphol. Embryol 1977, 23, 141–149. [Google Scholar]
- Bos, P.D.; Nguyen, D.X.; Massague, J. Modeling metastasis in the mouse. Curr. Opin. Pharmacol 2010, 10, 571–577. [Google Scholar]
- Khanna, C.; Hunter, K. Modeling metastasis in vivo. Carcinogenesis 2005, 26, 513–523. [Google Scholar]
- Cranmer, L.D.; Trevor, K.T.; Bandlamuri, S.; Hersh, E.M. Rodent models of brain metastasis in melanoma. Melanoma Res 2005, 15, 325–356. [Google Scholar]
- Fidler, I.J. Selection of successive tumour lines for metastasis. Nature 1973, 242, 148–149. [Google Scholar]
- Zhang, Z.; Hatori, T.; Nonaka, H. An experimental model of brain metastasis of lung carcinoma. Neuropathology 2008, 28, 24–28. [Google Scholar]
- Munoz, D.M.; Guha, A. Mouse models to interrogate the implications of the differentiation status in the ontogeny of gliomas. Oncotarget 2011, 2, 590–598. [Google Scholar]
- Mathieu, A.; Remmelink, M.; D’Haene, N.; Penant, S.; Gaussin, J.F.; van Ginckel, R.; Darro, F.; Kiss, R.; Salmon, I. Development of a chemoresistant orthotopic human nonsmall cell lung carcinoma model in nude mice: Analyses of tumor heterogenity in relation to the immunohistochemical levels of expression of cyclooxygenase-2, ornithine decarboxylase, lung-related resistance protein, prostaglandin E synthetase, and glutathione-S-transferase-α (GST)-α, GST-mu, and GST-pi. Cancer 2004, 101, 1908–1918. [Google Scholar]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar]
- Banisadr, G.; Skrzydelski, D.; Kitabgi, P.; Rostene, W.; Parsadaniantz, S.M. Highly regionalized distribution of stromal cell-derived factor-1/CXCL12 in adult rat brain: Constitutive expression in cholinergic, dopaminergic and vasopressinergic neurons. Eur. J. Neurosci 2003, 18, 1593–1606. [Google Scholar]
- Salmaggi, A.; Maderna, E.; Calatozzolo, C.; Gaviani, P.; Canazza, A.; Milanesi, I.; Silvani, A.; DiMeco, F.; Carbone, A.; Pollo, B. CXCL12, CXCR4 and CXCR7 expression in brain metastases. Cancer Biol. Ther 2009, 8, 1608–1614. [Google Scholar]
- Scala, S.; Ottaiano, A.; Ascierto, P.A.; Cavalli, M.; Simeone, E.; Giuliano, P.; Napolitano, M.; Franco, R.; Botti, G.; Castello, G. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. Clin. Cancer Res 2005, 11, 1835–1841. [Google Scholar]
- Schrader, A.J.; Lechner, O.; Templin, M.; Dittmar, K.E.; Machtens, S.; Mengel, M.; Probst-Kepper, M.; Franzke, A.; Wollensak, T.; Gatzlaff, P.; et al. CXCR4/CXCL12 expression and signalling in kidney cancer. Br. J. Cancer 2002, 86, 1250–1256. [Google Scholar]
- Zhou, Y.; Larsen, P.H.; Hao, C.; Yong, V.W. CXCR4 is a major chemokine receptor on glioma cells and mediates their survival. J. Biol. Chem 2002, 277, 49481–49487. [Google Scholar]
- Hinton, C.V.; Avraham, S.; Avraham, H.K. Role of the CXCR4/CXCL12 signaling axis in breast cancer metastasis to the brain. Clin. Exp. Metastasis 2010, 27, 97–105. [Google Scholar]
- Lee, B.C.; Lee, T.H.; Avraham, S.; Avraham, H.K. Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1α in breast cancer cell migration through human brain microvascular endothelial cells. Mol. Cancer Res 2004, 2, 327–338. [Google Scholar]
- Furusato, B.; Mohamed, A.; Uhlen, M.; Rhim, J.S. CXCR4 and cancer. Pathol. Int 2010, 60, 497–505. [Google Scholar]
- Naor, D.; Nedvetzki, S.; Golan, I.; Melnik, L.; Faitelson, Y. CD44 in cancer. Crit. Rev. Clin. Lab. Sci 2002, 39, 527–579. [Google Scholar]
- Misra, S.; Heldin, P.; Hascall, V.C.; Karamanos, N.K.; Skandalis, S.S.; Markwald, R.R.; Ghatak, S. Hyaluronan–CD44 interactions as potential targets for cancer therapy. FEBS J 2011, 278, 1429–1443. [Google Scholar]
- Sheridan, C.; Kishimoto, H.; Fuchs, R.K.; Mehrotra, S.; Bhat-Nakshatri, P.; Turner, C.H.; Goulet, R., Jr.; Badve, S.; Nakshatri, H. CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res 2006, 8, R59. [Google Scholar]
- Seiter, S.; Arch, R.; Reber, S.; Komitowski, D.; Hofmann, M.; Ponta, H.; Herrlich, P.; Matzku, S.; Zoller, M. Prevention of tumor metastasis formation by anti-variant CD44. J. Exp. Med 1993, 177, 443–455. [Google Scholar]
- Luo, Y.; Ziebell, M.R.; Prestwich, G.D. A hyaluronic acid-taxol antitumor bioconjugate targeted to cancer cells. Biomacromolecules 2000, 1, 208–218. [Google Scholar]
- Okuda, H.; Xing, F.; Pandey, P.R.; Sharma, S.; Watabe, M.; Pai, S.K.; Mo, Y.Y.; Iiizumi-Gairani, M.; Hirota, S.; Liu, Y.; et al. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res 2013, 73, 1434–1444. [Google Scholar]
- Xing, F.; Kobayashi, A.; Okuda, H.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Hirota, S.; Wilber, A.; Mo, Y.Y.; Moore, B.E.; et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol. Med 2013, 5, 384–396. [Google Scholar]
- Mendes, O.; Kim, H.T.; Lungu, G.; Stoica, G. MMP2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin. Exp. Metastasis 2007, 24, 341–351. [Google Scholar]
- Nguyen, D.X.; Chiang, A.C.; Zhang, X.H.; Kim, J.Y.; Kris, M.G.; Ladanyi, M.; Gerald, W.L.; Massague, J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 2009, 138, 51–62. [Google Scholar]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar]
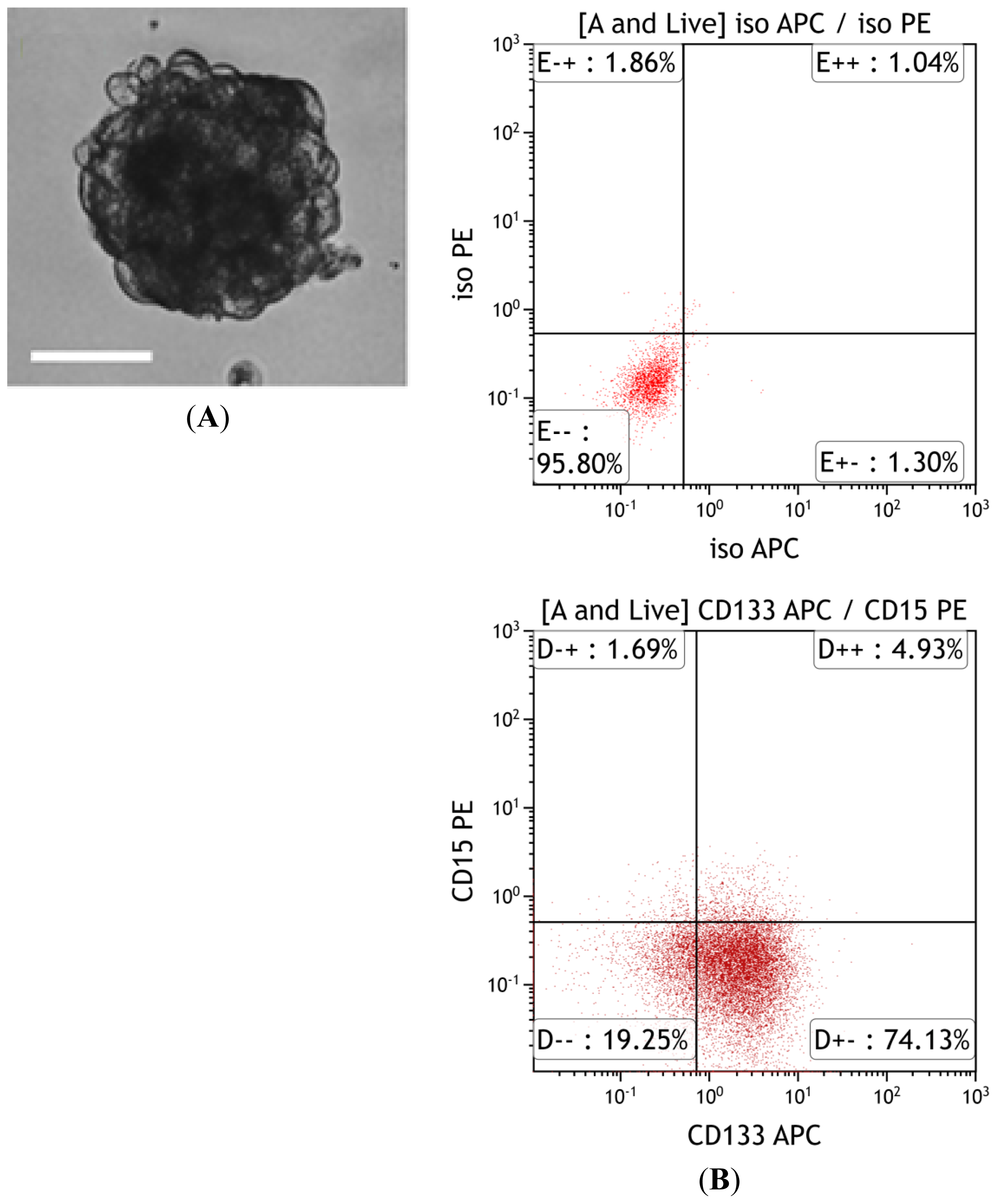

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Source | Incidence of BM | Metastastic Features |
|---|---|---|
| Lung | 40%–50% | Multiple metastatic lesions in the brain parenchyma in the early stages of the disease, and are associated with surrounding vasogenic edema |
| Breast | 15%–25% | Single lesions found in the parenchyma and leptomeninges, with rare occurrences of vasogenic edema |
| Skin (Melanoma) | 6%–11% | Multiple lesions form in the cortex as opposed to the grey-white junction, associated with hemorrhage |
| Colorectal | 3% | Lesions in the supratentorial and cerebellar regions |
| Unknown | 16% | Variable |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Singh, M.; Manoranjan, B.; Mahendram, S.; McFarlane, N.; Venugopal, C.; Singh, S.K. Brain Metastasis-Initiating Cells: Survival of the Fittest. Int. J. Mol. Sci. 2014, 15, 9117-9133. https://doi.org/10.3390/ijms15059117
Singh M, Manoranjan B, Mahendram S, McFarlane N, Venugopal C, Singh SK. Brain Metastasis-Initiating Cells: Survival of the Fittest. International Journal of Molecular Sciences. 2014; 15(5):9117-9133. https://doi.org/10.3390/ijms15059117
Chicago/Turabian StyleSingh, Mohini, Branavan Manoranjan, Sujeivan Mahendram, Nicole McFarlane, Chitra Venugopal, and Sheila K. Singh. 2014. "Brain Metastasis-Initiating Cells: Survival of the Fittest" International Journal of Molecular Sciences 15, no. 5: 9117-9133. https://doi.org/10.3390/ijms15059117