Molecular Biology of Brain Metastasis

{kind=link}
Abstract
:1. Introduction
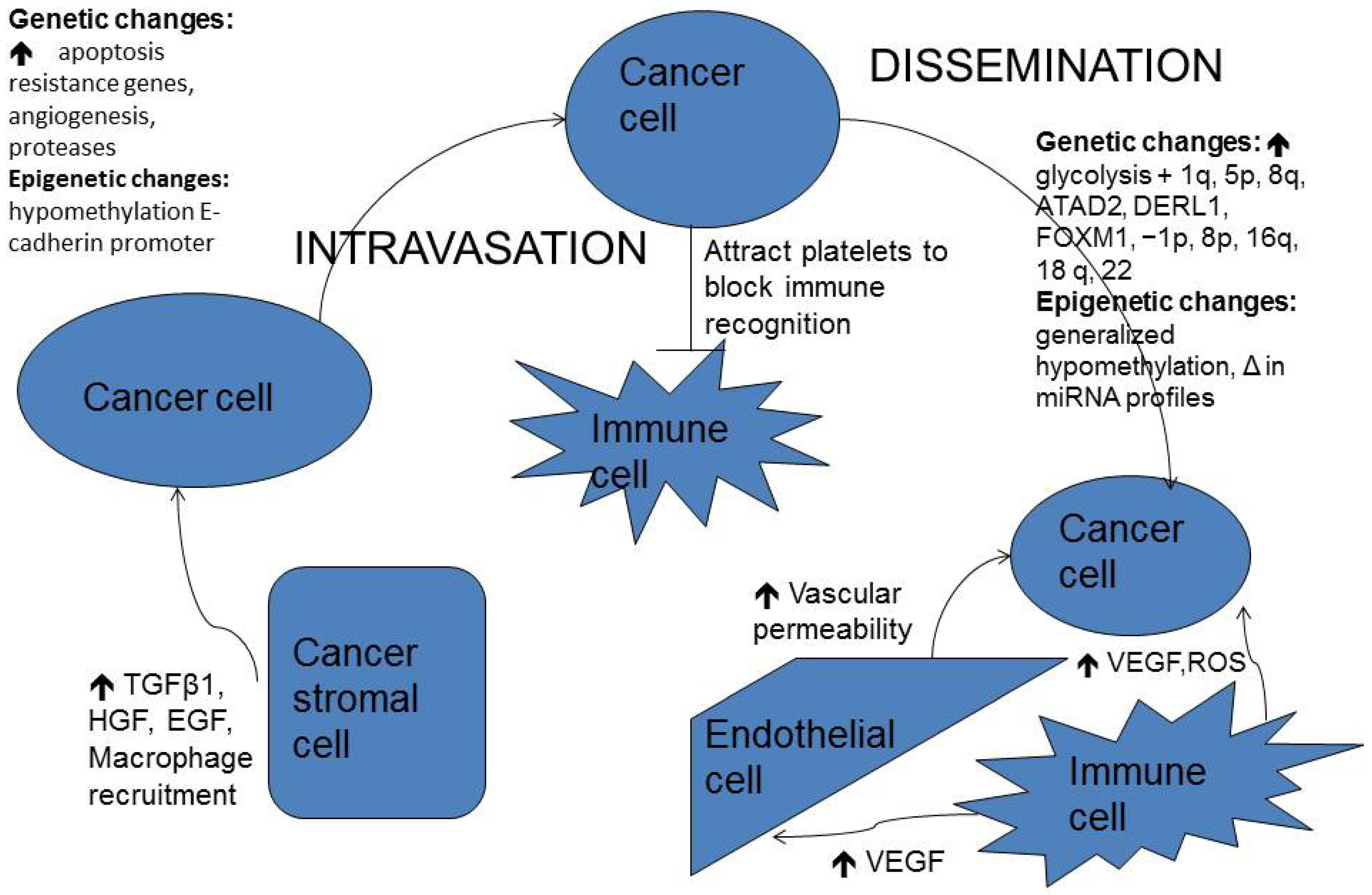
2. Pathogenesis of Brain Metastasis
2.1. Cellular Heterogeneity and Proliferation
2.2. Epithelial-Mesenchymal Transition (EMT)
2.3. Interaction with Tumor Stroma
2.4. Local Invasion
2.5. E-Cadherin-Catenin Complex and Integrins
2.6. Dissemination
2.7. The Brain’s Microenvironment
2.8. Contribution of Systemic Immune Cells to Metastasis Proliferation on the Brain
3. Evolution of Brain Metastasis Animal Models
3.1. Current Brain Metastases Models
3.2. Genetics in Breast Cancer Brain Metastases
4. Epigenetics in Brain Metastases
4.1. DNA Methylation
4.2. MicroRNAs in Brain Metastases

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barnholtz-Sloan, J.S.; Sloan, A.E.; Davis, F.G.; Vigneau, F.D.; Lai, P.; Sawaya, R.E. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the metropolitan detroit cancer surveillance system. J. Clin. Oncol. 2004, 22, 2865–2872. [Google Scholar] [CrossRef]
- McDermott, R.; Gabikian, P.; Sarvaiya, P.; Ulasov, I.; Lesniak, M.S. Micrornas in brain metastases: Big things come in small packages. J. Mol. Med. 2013, 91, 5–13. [Google Scholar] [CrossRef]
- Barajas, R.F., Jr.; Cha, S. Imaging diagnosis of brain metastasis. Prog. Neurol. Surg. 2012, 25, 55–73. [Google Scholar] [CrossRef]
- Delattre, J.Y.; Krol, G.; Thaler, H.T.; Posner, J.B. Distribution of brain metastases. Arch. Neurol. 1988, 45, 741–744. [Google Scholar] [CrossRef]
- Chang, E.L.; Wefel, J.S.; Maor, M.H.; Hassenbusch, S.J., 3rd; Mahajan, A.; Lang, F.F.; Woo, S.Y.; Mathews, L.A.; Allen, P.K.; Shiu, A.S.; et al. A pilot study of neurocognitive function in patients with one to three new brain metastases initially treated with stereotactic radiosurgery alone. Neurosurgery 2007, 60, 277–283. [Google Scholar]
- Lockman, P.R.; Mittapalli, R.K.; Taskar, K.S.; Rudraraju, V.; Gril, B.; Bohn, K.A.; Adkins, C.E.; Roberts, A.; Thorsheim, H.R.; Gaasch, J.A.; et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 2010, 16, 5664–5678. [Google Scholar] [CrossRef]
- Beasley, K.D.; Toms, S.A. The molecular pathobiology of metastasis to the brain: A review. Neurosurg. Clin. N. Am. 2011, 22, 7–14. [Google Scholar] [CrossRef]
- Junttila, M.R.; Evan, G.I. P53—A jack of all trades but master of none. Nat. Rev. Cancer 2009, 9, 821–829. [Google Scholar] [CrossRef]
- Lyden, D.; Hattori, K.; Dias, S.; Costa, C.; Blaikie, P.; Butros, L.; Chadburn, A.; Heissig, B.; Marks, W.; Witte, L.; et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 2001, 7, 1194–1201. [Google Scholar] [CrossRef]
- Ailles, L.E.; Weissman, I.L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol. 2007, 18, 460–466. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Pan, D.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar] [CrossRef]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Hirohashi, S.; Kanai, Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci. 2003, 94, 575–581. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Veve, R.; Hirsch, F.R.; Franklin, W.A. The e-cadherin cell-cell adhesion complex and lung cancer invasion, metastasis, and prognosis. Lung Cancer 2002, 36, 115–124. [Google Scholar] [CrossRef]
- Hulit, J.; Suyama, K.; Chung, S.; Keren, R.; Agiostratidou, G.; Shan, W.; Dong, X.; Williams, T.M.; Lisanti, M.P.; Knudsen, K.; et al. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 2007, 67, 3106–3116. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef]
- Marchetti, D.; Nicolson, G.L. Human heparanase: A molecular determinant of brain metastasis. Adv. Enzym. Regul. 2001, 41, 343–359. [Google Scholar] [CrossRef]
- Davies, P.F.; Spaan, J.A.; Krams, R. Shear stress biology of the endothelium. Ann. Biomed. Eng. 2005, 33, 1714–1718. [Google Scholar] [CrossRef]
- Kumar, S.; Weaver, V.M. Mechanics, malignancy, and metastasis: The force journey of a tumor cell. Cancer Metastasis Rev. 2009, 28, 113–127. [Google Scholar] [CrossRef]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Mannel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar]
- Weil, R.J.; Palmieri, D.C.; Bronder, J.L.; Stark, A.M.; Steeg, P.S. Breast cancer metastasis to the central nervous system. Am. J. Pathol. 2005, 167, 913–920. [Google Scholar] [CrossRef]
- Lesniak, M.S.; Brem, H. Targeted therapy for brain tumours. Nat. Rev. Drug Discov. 2004, 3, 499–508. [Google Scholar] [CrossRef]
- Lorger, M. Tumor microenvironment in the brain. Cancers 2012, 4, 218–243. [Google Scholar] [CrossRef]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R., 3rd; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef]
- Raichle, M.E.; Mintun, M.A. Brain work and brain imaging. Annu. Rev. Neurosci. 2006, 29, 449–476. [Google Scholar] [CrossRef]
- Rao, J.; Oz, G.; Seaquist, E.R. Regulation of cerebral glucose metabolism. Minerva Endocr. 2006, 31, 149–158. [Google Scholar]
- Green, C.E.; Liu, T.; Montel, V.; Hsiao, G.; Lester, R.D.; Subramaniam, S.; Gonias, S.L.; Klemke, R.L. Chemoattractant signaling between tumor cells and macrophages regulates cancer cell migration, metastasis and neovascularization. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Haddad, O.; Chotard-Ghodsnia, R.; Verdier, C.; Duperray, A. Tumor cell/endothelial cell tight contact upregulates endothelial adhesion molecule expression mediated by nfkappab: Differential role of the shear stress. Exp. Cell Res. 2010, 316, 615–626. [Google Scholar]
- Munzarova, M.; Kovarik, J. Is cancer a macrophage-mediated autoaggressive disease? Lancet 1987, 1, 952–954. [Google Scholar] [CrossRef]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef]
- Maher, E.A.; Mietz, J.; Arteaga, C.L.; DePinho, R.A.; Mohla, S. Brain metastasis: Opportunities in basic and translational research. Cancer Res. 2009, 69, 6015–6020. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Kouros-Mehr, H.; Bechis, S.K.; Slorach, E.M.; Littlepage, L.E.; Egeblad, M.; Ewald, A.J.; Pai, S.Y.; Ho, I.C.; Werb, Z. Gata-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell 2008, 13, 141–152. [Google Scholar] [CrossRef]
- Lee, B.C.; Lee, T.H.; Avraham, S.; Avraham, H.K. Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1alpha in breast cancer cell migration through human brain microvascular endothelial cells. Mol. Cancer Res. 2004, 2, 327–338. [Google Scholar]
- Perou, C.M.; Parker, J.S.; Prat, A.; Ellis, M.J.; Bernard, P.S. Clinical implementation of the intrinsic subtypes of breast cancer. Lancet Oncol. 2010, 11, 718–719. [Google Scholar]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Nikolsky, Y.; Sviridov, E.; Yao, J.; Dosymbekov, D.; Ustyansky, V.; Kaznacheev, V.; Dezso, Z.; Mulvey, L.; Macconaill, L.E.; Winckler, W.; et al. Genome-wide functional synergy between amplified and mutated genes in human breast cancer. Cancer Res. 2008, 68, 9532–9540. [Google Scholar] [CrossRef]
- Yao, J.; Weremowicz, S.; Feng, B.; Gentleman, R.C.; Marks, J.R.; Gelman, R.; Brennan, C.; Polyak, K. Combined cdna array comparative genomic hybridization and serial analysis of gene expression analysis of breast tumor progression. Cancer Res. 2006, 66, 4065–4078. [Google Scholar] [CrossRef]
- Salhia, B.; Kiefer, J.; Ross, J.T.; Metapally, R.; Martinez, R.A.; Johnson, K.N.; DiPerna, D.M.; Paquette, K.M.; Jung, S.; Nasser, S.; et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Raeder, M.B.; Birkeland, E.; Trovik, J.; Krakstad, C.; Shehata, S.; Schumacher, S.; Zack, T.I.; Krohn, A.; Werner, H.M.; Moody, S.E.; et al. Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to myc-dependent cancers. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microrna targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Svokos, K.A.; Salhia, B.; Toms, S.A. Molecular Biology of Brain Metastasis. Int. J. Mol. Sci. 2014, 15, 9519-9530. https://doi.org/10.3390/ijms15069519
Svokos KA, Salhia B, Toms SA. Molecular Biology of Brain Metastasis. International Journal of Molecular Sciences. 2014; 15(6):9519-9530. https://doi.org/10.3390/ijms15069519
Chicago/Turabian StyleSvokos, Konstantina A., Bodour Salhia, and Steven A. Toms. 2014. "Molecular Biology of Brain Metastasis" International Journal of Molecular Sciences 15, no. 6: 9519-9530. https://doi.org/10.3390/ijms15069519