Ginsenoside Rd Attenuates Mitochondrial Permeability Transition and Cytochrome c Release in Isolated Spinal Cord Mitochondria: Involvement of Kinase-Mediated Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
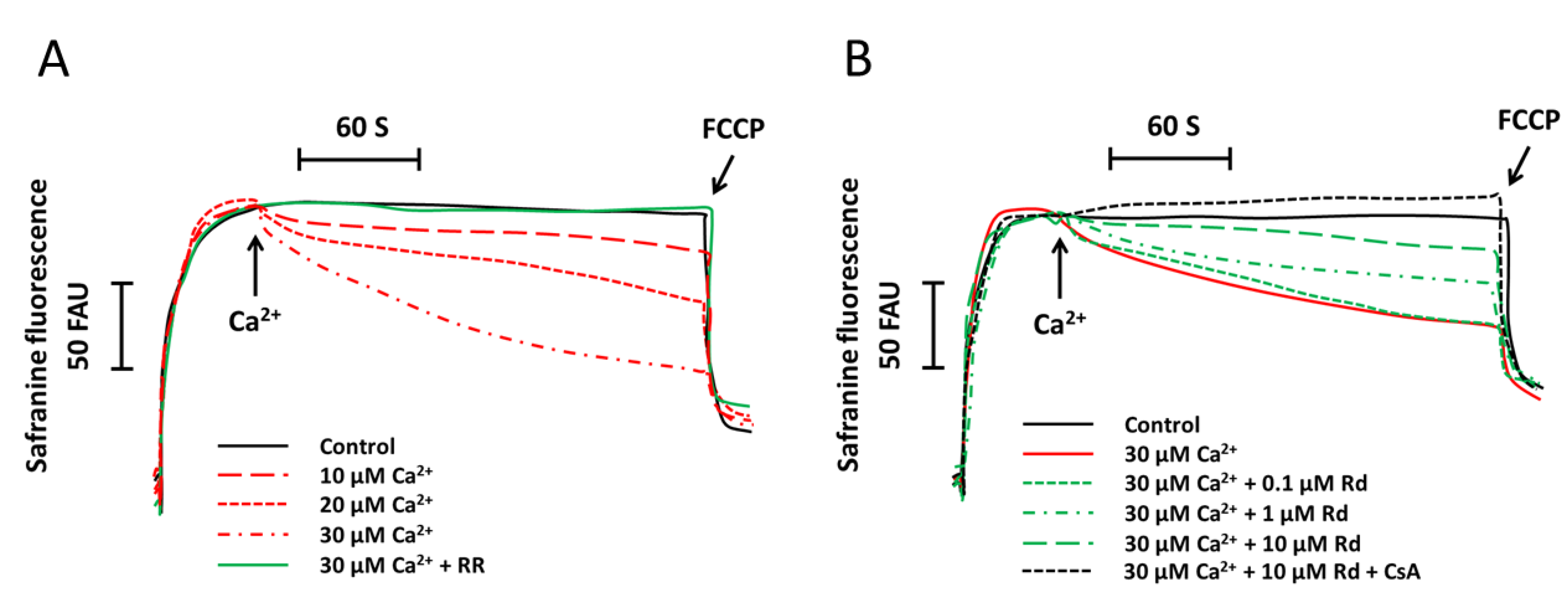
2.1. Rd (Ginsenoside Rd) Protects Isolated Spinal Cord Mitochondria against Ca2+ Induced Mitochondrial Membrane Potential Dissipation
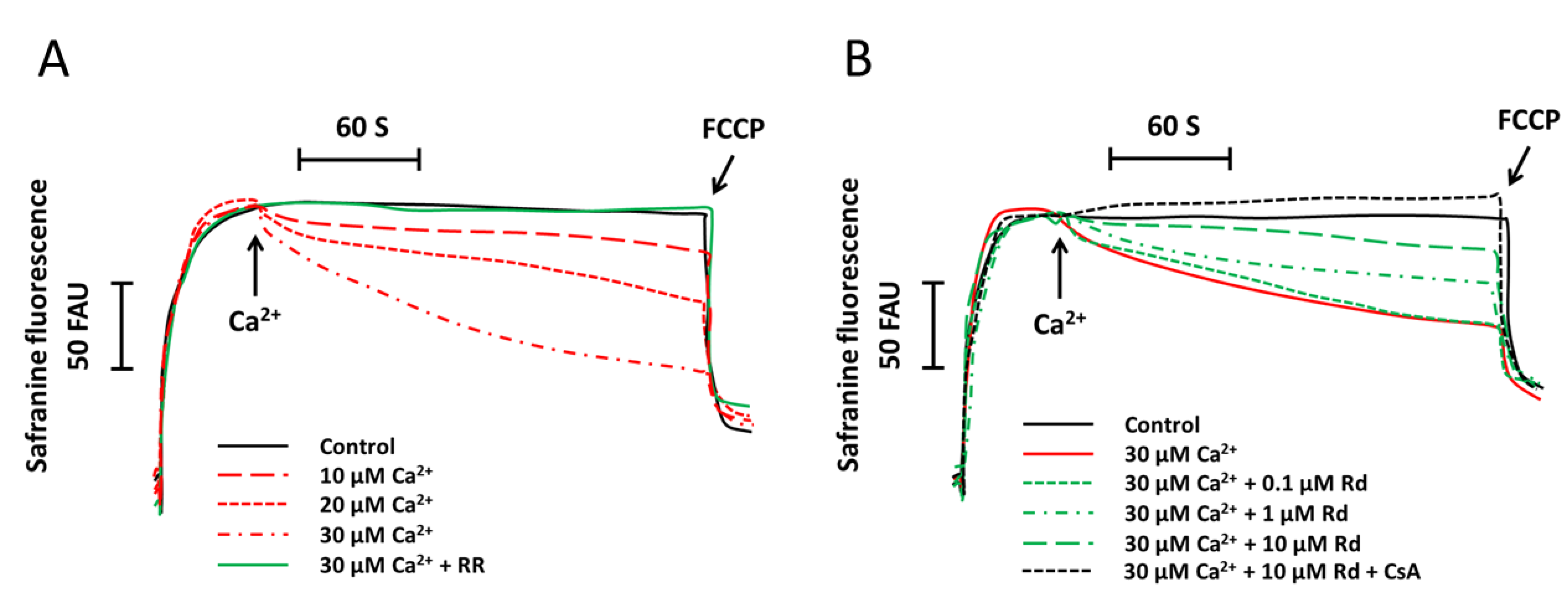
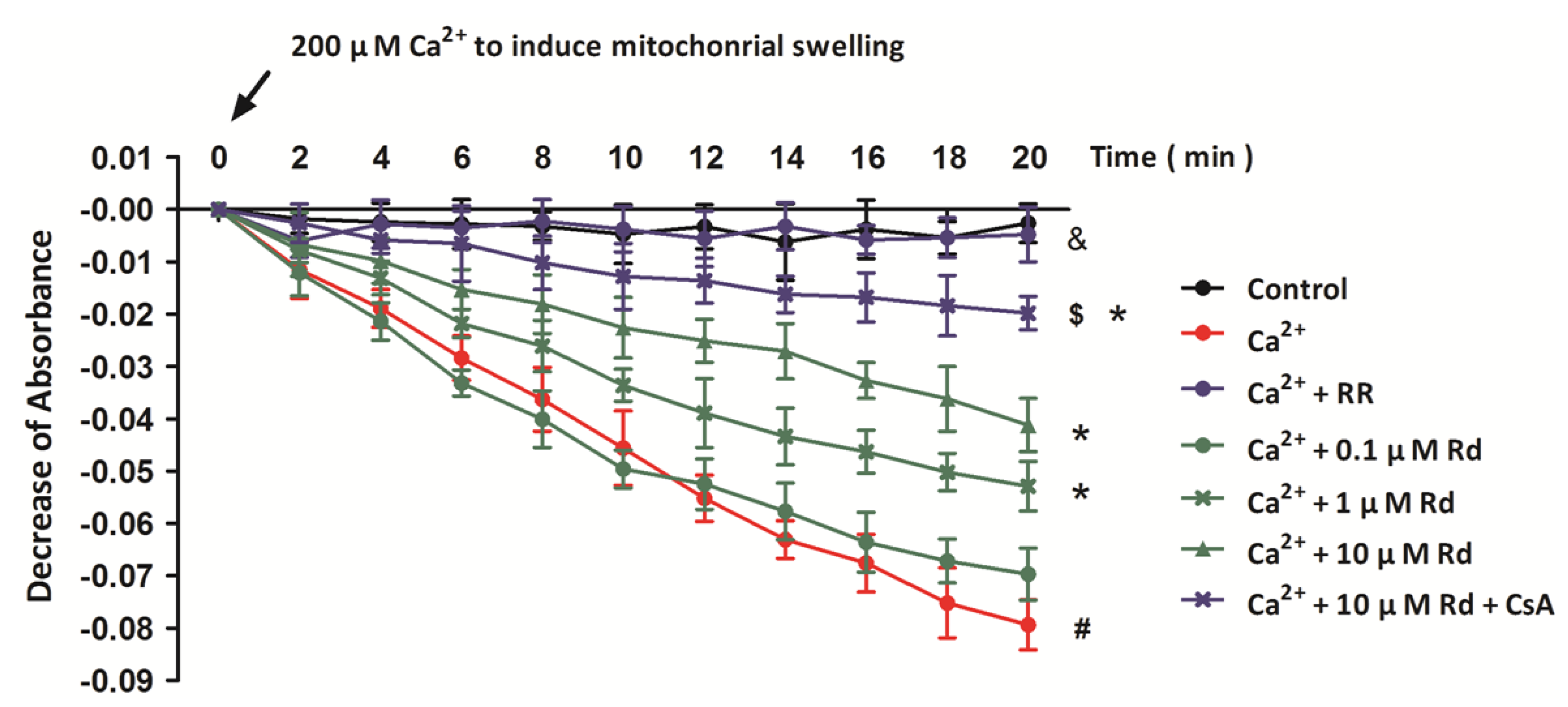
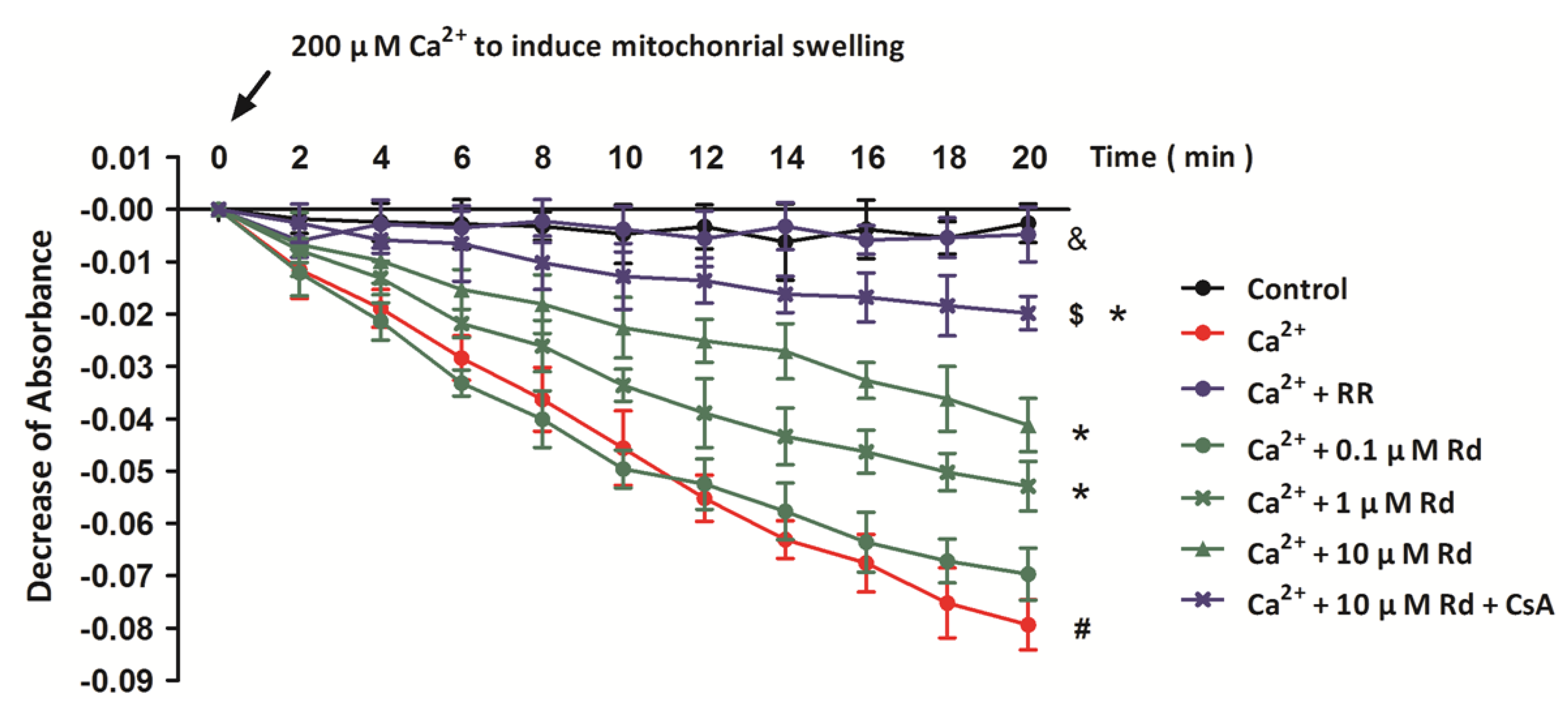
2.2. Rd Protects Isolated Spinal Cord Mitochondria against Ca2+ Induced Mitochondrial Swelling
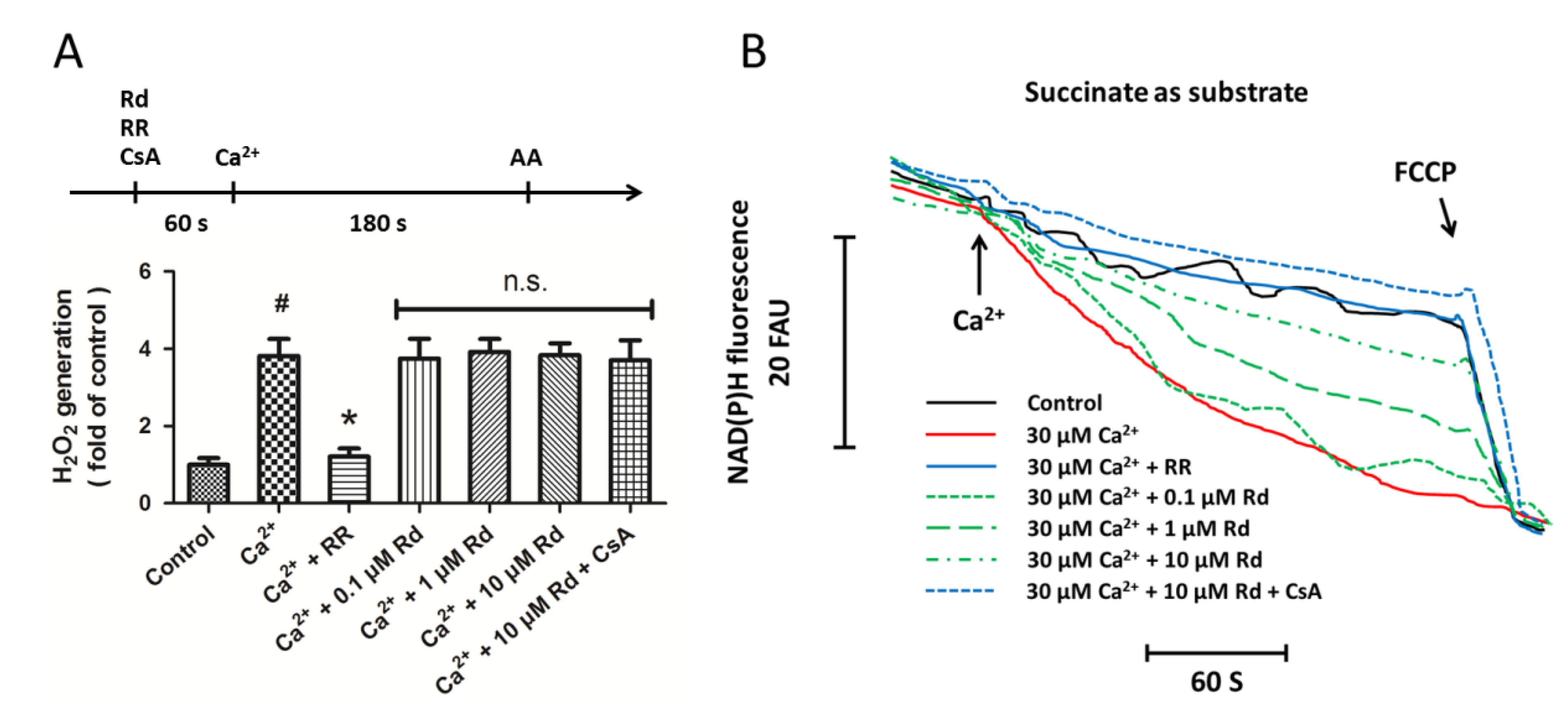
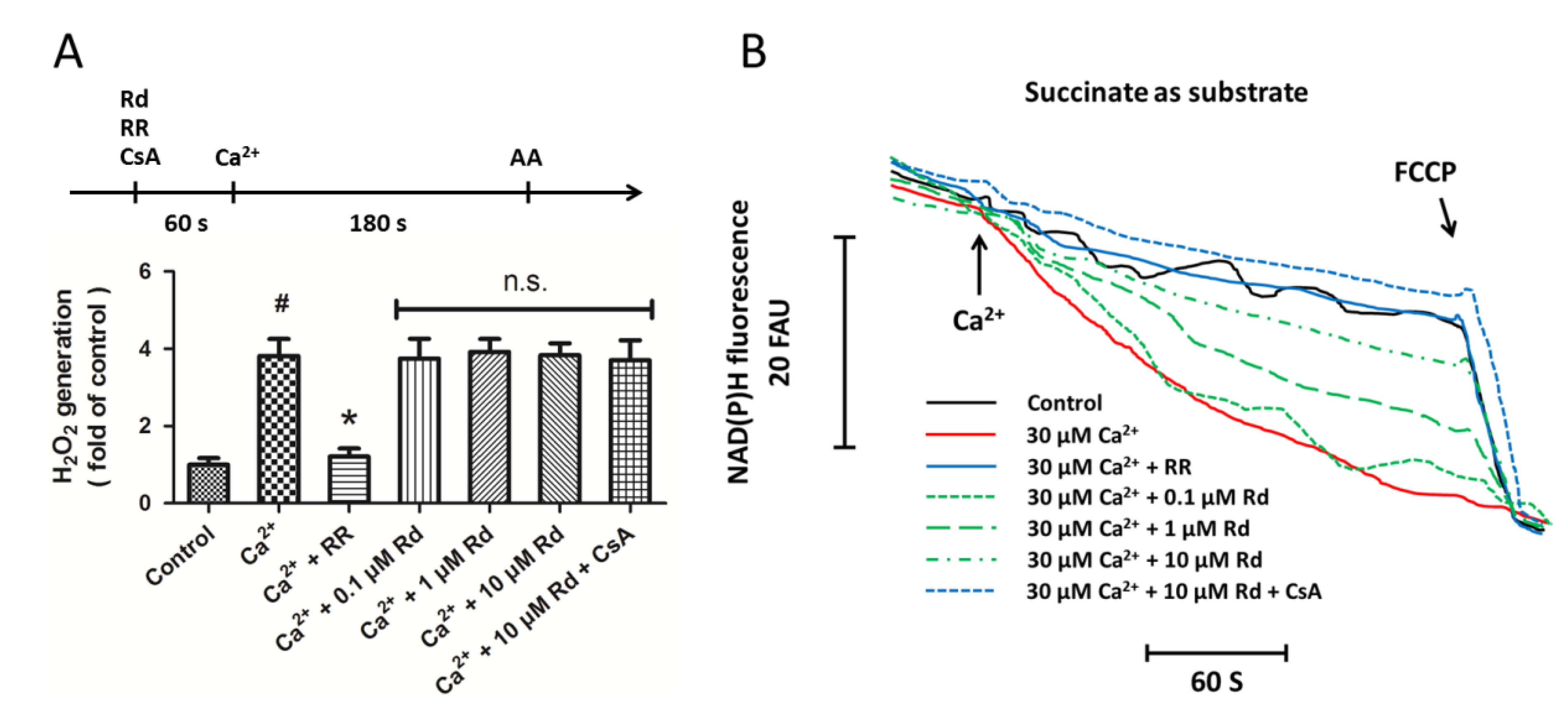
2.3. Effects of Rd on Mitochondrial H2O2 Generation and NAD(P)H Content after Ca2+ Treatment in Isolated Spinal Cord Mitochondria
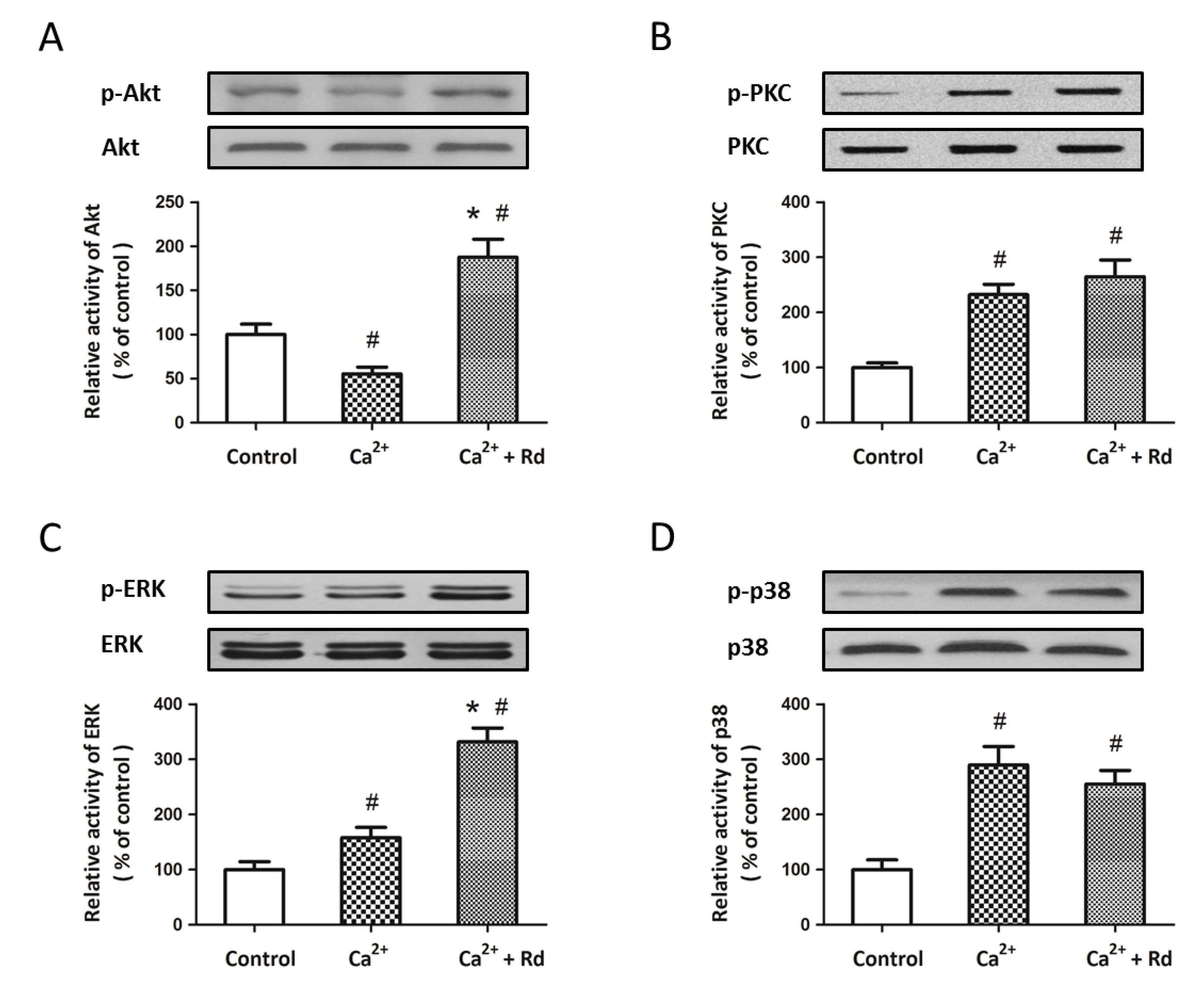
2.4. Effects of Rd on the Phosphorylation of Mitochondrial Protein Kinases after Ca2+ Treatment in Isolated Spinal Cord Mitochondria

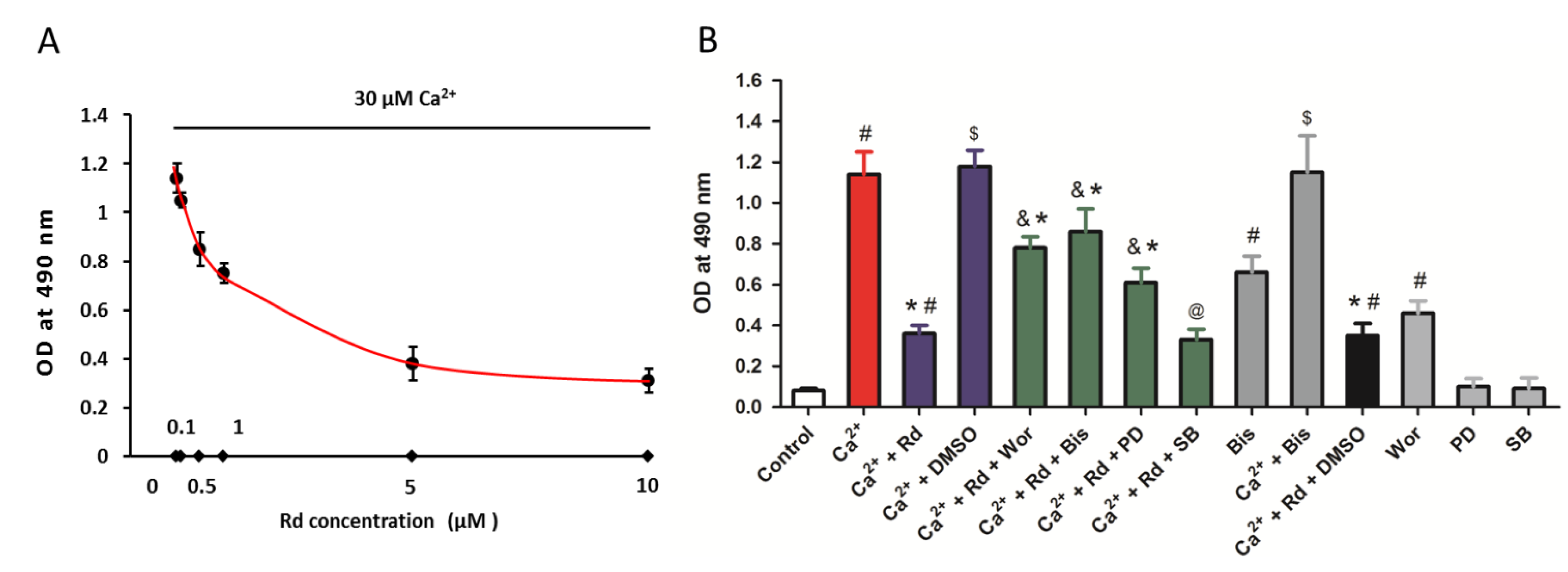
2.5. Effects of Rd and Protein Kinase Inhibitors on Cytochrome c Release after Ca2+ Treatment in Isolated Spinal Cord Mitochondria

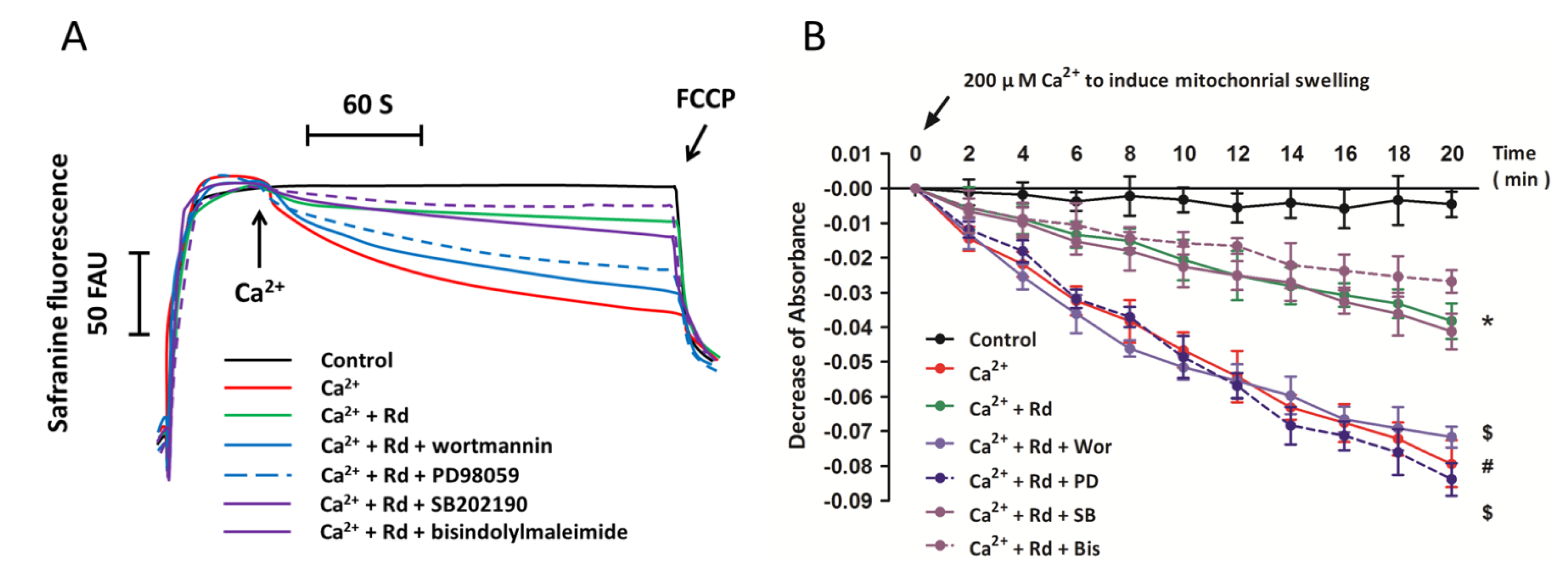
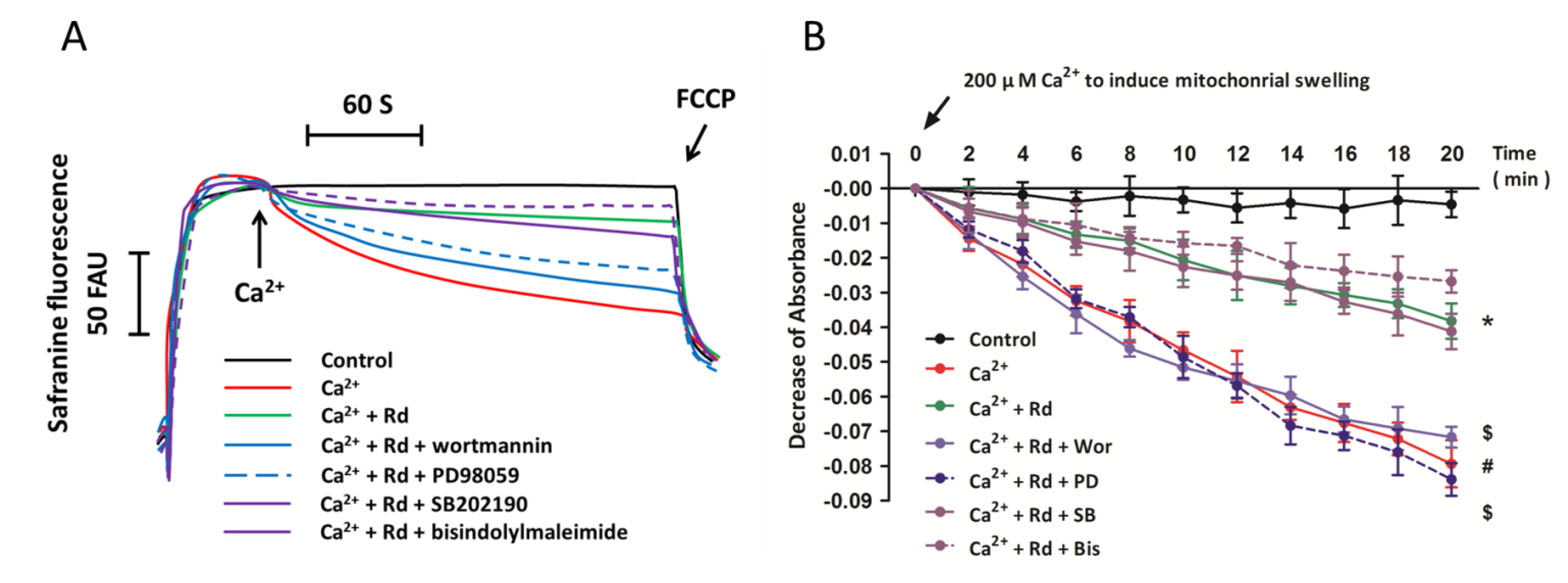
2.6. Effects of Protein Kinase Inhibitors on Mitochondrial Membrane Potential Dissipation and Swelling after Ca2+ Treatment in Rd Pretreated Spinal Cord Mitochondria
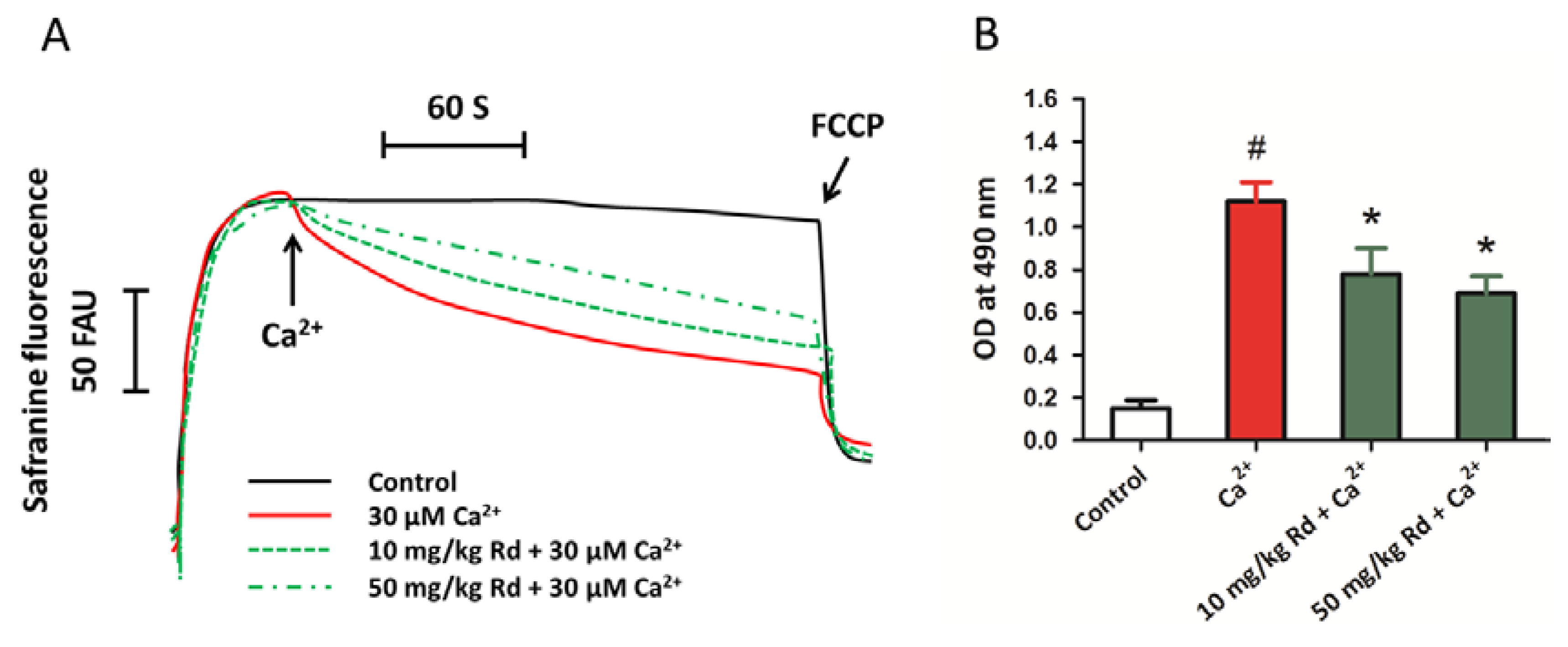
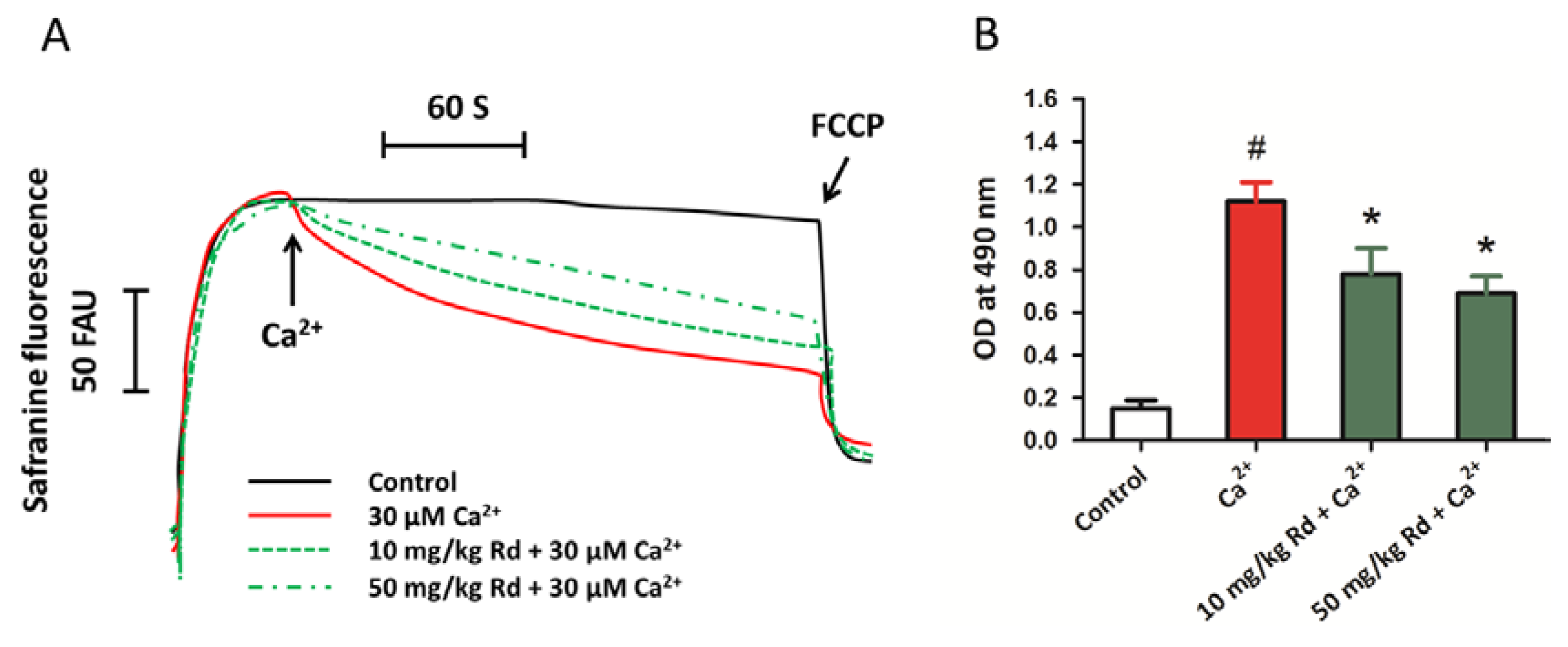
2.7. Protective Activity of Rd against Mitochondrial Permeability Transition and Cytochrome c Release through in Vivo Administration
2.8. Discussion
3. Experimental Section
3.1. Animals
3.2. Mitochondria Purification
3.3. Rd Treatment
3.4. Mitochondrial Membrane Potential Measurement
3.5. Determination of Mitochondrial Swelling
3.6. Measurement of H2O2 Production
3.7. Measurement of NAD(P)H Content
3.8. Western Blot Analysis
3.9. Determination of Cytochrome c Release
3.10. Statistical Analysis
4. Conclusions
Conflicts of Interest
References
- Svensson, L.G.; Crawford, E.S.; Hess, K.R.; Coselli, J.S.; Safi, H.J. Experience with 1509 patients undergoing thoracoabdominal aortic operations. J. Vasc. Surg. 1993, 17, 357–368. [Google Scholar] [CrossRef]
- Zurita, M.; Aguayo, C.; Bonilla, C.; Otero, L.; Rico, M.; Rodriguez, A.; Vaquero, J. The pig model of chronic paraplegia: A challenge for experimental studies in spinal cord injury. Prog. Neurobiol. 2012, 97, 288–303. [Google Scholar]
- Chiesa, R.; Melissano, G.; Civilini, E.; de Moura, M.L.; Carozzo, A.; Zangrillo, A. Ten years experience of thoracic and thoracoabdominal aortic aneurysm surgical repair: lessons learned. Ann. Vasc. Surg. 2004, 18, 514–520. [Google Scholar] [CrossRef]
- Etz, C.D.; Homann, T.M.; Luehr, M.; Kari, F.A.; Weisz, D.J.; Kleinman, G.; Plestis, K.A.; Griepp, R.B. Spinal cord blood flow and ischemic injury after experimental sacrifice of thoracic and abdominal segmental arteries. Eur. J. Cardiothorac. Surg. 2008, 33, 1030–1038. [Google Scholar] [CrossRef]
- Chang, C.K.; Chuter, T.A.; Reilly, L.M.; Ota, M.K.; Furtado, A.; Bucci, M.; Wintermark, M.; Hiramoto, J.S. Spinal arterial anatomy and risk factors for lower extremity weakness following endovascular thoracoabdominal aortic aneurysm repair with branched stent-grafts. J. Endovasc. Ther. 2008, 15, 356–362. [Google Scholar] [CrossRef]
- Backes, W.H.; Nijenhuis, R.J.; Mess, W.H.; Wilmink, F.A.; Schurink, G.W.; Jacobs, M.J. Magnetic resonance angiography of collateral blood supply to spinal cord in thoracic and thoracoabdominal aortic aneurysm patients. J. Vasc. Surg. 2008, 48, 261–271. [Google Scholar] [CrossRef]
- Ogino, H.; Sasaki, H.; Minatoya, K.; Matsuda, H.; Yamada, N.; Kitamura, S. Combined use of adamkiewicz artery demonstration and motor-evoked potentials in descending and thoracoabdominal repair. Ann. Thorac. Surg. 2006, 82, 592–596. [Google Scholar] [CrossRef]
- Jacobs, M.J.; Mess, W.; Mochtar, B.; Nijenhuis, R.J.; Statius van Eps, R.G.; Schurink, G.W. The value of motor evoked potentials in reducing paraplegia during thoracoabdominal aneurysm repair. J. Vasc. Surg. 2006, 43, 239–246. [Google Scholar] [CrossRef]
- Melissano, G.; Chiesa, R. Advances in imaging of the spinal cord vascular supply and its relationship with paraplegia after aortic interventions. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 567–577. [Google Scholar] [CrossRef]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Neuroscience 2004, 5, 146–156. [Google Scholar]
- Saxena, P.; Abu Hasan, F.; Merry, C. Spinal cord ischemia following coronary artery bypass surgery. J. Card. Surg. 2012, 27, 45–46. [Google Scholar] [CrossRef]
- De Haan, P.; Kalkman, C.J.; Jacobs, M.J. Pharmacologic neuroprotection in experimental spinal cord ischemia: A systematic review. J. Neurosurg. Anesthesiol. 2001, 13, 3–12. [Google Scholar] [CrossRef]
- Ahn, Y.H.; Lee, G.; Kang, S.K. Molecular insights of the injured lesions of rat spinal cords: Inflammation, apoptosis, and cell survival. Biochem. Biophys. Res. Commun. 2006, 348, 560–570. [Google Scholar] [CrossRef]
- Genovese, T.; Cuzzocrea, S. Role of free radicals and poly (ADP-ribose) polymerase-1 in the development of spinal cord injury: New potential therapeutic targets. Curr. Med. Chem. 2008, 15, 477–487. [Google Scholar] [CrossRef]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de Leon, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Li, L.; Feng, F.; Yuan, H.; Gao, D.K.; Fu, L.A.; Fei, Z. Osthole attenuates spinal cord ischemia-reperfusion injury through mitochondrial biogenesis-independent inhibition of mitochondrial dysfunction in rats. J. Surg. Res. 2013, 185, 805–814. [Google Scholar] [CrossRef]
- Zhu, J.W.; Chen, T.; Guan, J.; Liu, W.B.; Liu, J. Neuroprotective effects of allicin on spinal cord ischemia-reperfusion injury via improvement of mitochondrial function in rabbits. Neurochem. Int. 2012, 61, 640–648. [Google Scholar] [CrossRef]
- Fu, J.; Fan, H.B.; Guo, Z.; Wang, Z.; Li, X.D.; Li, J.; Pei, G.X. Salvianolic acid B attenuates spinal cord ischemia-reperfusion-induced neuronal injury and oxidative stress by activating the extracellular signal-regulated kinase pathway in rats. J. Surg. Res. 2014, 188, 222–230. [Google Scholar] [CrossRef]
- Jia, L.; Zhao, Y. Current evaluation of the millennium phytomedicine-ginseng (I): Etymology, pharmacognosy, phytochemistry, market and regulations. Curr. Med. Chem. 2009, 16, 2475–2484. [Google Scholar] [CrossRef]
- Peng, L.; Sun, S.; Xie, L.H.; Wicks, S.M.; Xie, J.T. Ginsenoside Re: Pharmacological effects on cardiovascular system. Cardiovasc. Ther. 2012, 30, 183–188. [Google Scholar] [CrossRef]
- Lu, J.M.; Yao, Q.; Chen, C. Ginseng compounds: An update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef]
- Christensen, L.P.; Jensen, M. Biomass and content of ginsenosides and polyacetylenes in American ginseng roots can be increased without affecting the profile of bioactive compounds. J. Nat. Med. 2009, 63, 159–168. [Google Scholar] [CrossRef]
- Christensen, L.P. Ginsenosides chemistry, biosynthesis, analysis, and potential health effects. Adv. Food Nutr. Res. 2009, 55, 1–99. [Google Scholar] [CrossRef]
- Ye, R.; Zhao, G.; Liu, X. Ginsenoside Rd for acute ischemic stroke: translating from bench to bedside. Expert Rev. Neurother. 2013, 13, 603–613. [Google Scholar] [CrossRef]
- Zhang, C.; Du, F.; Shi, M.; Ye, R.; Cheng, H.; Han, J.; Ma, L.; Cao, R.; Rao, Z.; Zhao, G. Ginsenoside Rd protects neurons against glutamate-induced excitotoxicity by inhibiting Ca2+ influx. Cell. Mol. Neurobiol. 2012, 32, 121–128. [Google Scholar] [CrossRef]
- Ye, R.; Han, J.; Kong, X.; Zhao, L.; Cao, R.; Rao, Z.; Zhao, G. Protective effects of ginsenoside Rd on PC12 cells against hydrogen peroxide. Biol. Pharm. Bull. 2008, 31, 1923–1927. [Google Scholar] [CrossRef]
- Ye, R.; Li, N.; Han, J.; Kong, X.; Cao, R.; Rao, Z.; Zhao, G. Neuroprotective effects of ginsenoside Rd against oxygen-glucose deprivation in cultured hippocampal neurons. Neurosci. Res. 2009, 64, 306–310. [Google Scholar] [CrossRef]
- Ye, R.; Kong, X.; Yang, Q.; Zhang, Y.; Han, J.; Zhao, G. Ginsenoside Rd attenuates redox imbalance and improves stroke outcome after focal cerebral ischemia in aged mice. Neuropharmacology 2011, 61, 815–824. [Google Scholar] [CrossRef]
- Ye, R.; Kong, X.; Yang, Q.; Zhang, Y.; Han, J.; Li, P.; Xiong, L.; Zhao, G. Ginsenoside rd in experimental stroke: Superior neuroprotective efficacy with a wide therapeutic window. Neurotherapeutics 2011, 8, 515–525. [Google Scholar] [CrossRef]
- Ye, R.; Zhang, X.; Kong, X.; Han, J.; Yang, Q.; Zhang, Y.; Chen, Y.; Li, P.; Liu, J.; Shi, M.; et al. Ginsenoside Rd attenuates mitochondrial dysfunction and sequential apoptosis after transient focal ischemia. Neuroscience 2011, 178, 169–180. [Google Scholar] [CrossRef]
- Ye, R.; Yang, Q.; Kong, X.; Han, J.; Zhang, X.; Zhang, Y.; Li, P.; Liu, J.; Shi, M.; Xiong, L.; Zhao, G. Ginsenoside Rd attenuates early oxidative damage and sequential inflammatory response after transient focal ischemia in rats. Neurochem. Int. 2011, 58, 391–398. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Dubinsky, J.M. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J. Neurosci. 2000, 20, 8229–8237. [Google Scholar]
- Kowalczyk, J.E.; Zablocka, B. [Protein kinases in mitochondria]. Postep. Biochem. 2008, 54, 209–216. [Google Scholar]
- Antico Arciuch, V.G.; Alippe, Y.; Carreras, M.C.; Poderoso, J.J. Mitochondrial kinases in cell signaling: Facts and perspectives. Adv. Drug Deliv. Rev. 2009, 61, 1234–1249. [Google Scholar] [CrossRef]
- Lopez, D.; Fischbach, M.A.; Chu, F.; Losick, R.; Kolter, R. Structurally diverse natural products that cause potassium leakage trigger multicellularity in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2009, 106, 280–285. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wang, X. Recent advances in natural products from plants for treatment of liver diseases. Eur. J. Med. Chem. 2013, 63, 570–577. [Google Scholar] [CrossRef]
- McEwen, M.L.; Sullivan, P.G.; Rabchevsky, A.G.; Springer, J.E. Targeting mitochondrial function for the treatment of acute spinal cord injury. Neurotherapeutics 2011, 8, 168–179. [Google Scholar] [CrossRef]
- Shi, P.; Wei, Y.; Zhang, J.; Gal, J.; Zhu, H. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J. Alzheimer’s Dis. 2010, 20, S311–S324. [Google Scholar]
- Sun, D.; Wang, B.; Shi, M.; Zhang, Y.X.; Zhou, L.F.; Liu, Z.R.; Wu, Z.L.; Jiang, W.; Han, J.L.; Xiong, L.Z.; et al. Pharmacokinetic, tissue distribution and excretion of ginsenoside-Rd in rodents. Phytomedicine 2012, 19, 369–373. [Google Scholar] [CrossRef]
- Wang, W.; Wang, G.J.; Xie, H.T.; Sun, J.G.; Zhao, S.; Jiang, X.L.; Li, H.; Lv, H.; Xu, M.J.; Wang, R. Determination of ginsenoside Rd in dog plasma by liquid chromatography-mass spectrometry after solid-phase extraction and its application in dog pharmacokinetics studies. J. Chromatogr. B 2007, 852, 8–14. [Google Scholar] [CrossRef]
- Deng, Y.H.; Feng, Y.; Zeng, X.; Yang, L.; Liu, Y.M.; Huang, Y.; Sun, J.; Zhou, D.; Liang, W.X. Determination of ginsenoside rd in human plasma by LC/MS/MS. Zhong yao cai/Zhongyaocai/J. Chin. Med. Mater. 2006, 29, 928–931. (In Chinese) [Google Scholar]
- Liu, X.; Xia, J.; Wang, L.; Song, Y.; Yang, J.; Yan, Y.; Ren, H.; Zhao, G. Efficacy and safety of ginsenoside-Rd for acute ischaemic stroke: A randomized, double-blind, placebo-controlled, phase II multicenter trial. Eur. J. Neurol. 2009, 16, 569–575. [Google Scholar] [CrossRef]
- Moore, C.L. Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochem. Biophys. Res. Commun. 1971, 42, 298–305. [Google Scholar] [CrossRef]
- Bernardi, P.; von Stockum, S. The permeability transition pore as a Ca2+ release channel: New answers to an old question. Cell Calcium 2012, 52, 22–27. [Google Scholar] [CrossRef]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef]
- Armogida, M.; Nistico, R.; Mercuri, N.B. Therapeutic potential of targeting hydrogen peroxide metabolism in the treatment of brain ischaemia. Br. J. Pharmacol. 2012, 166, 1211–1224. [Google Scholar] [CrossRef]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Bijur, G.N.; Jope, R.S. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003, 87, 1427–1435. [Google Scholar] [CrossRef]
- Galli, S.; Arciuch, V.G.A.; Poderoso, C.; Converso, D.P.; Zhou, Q.; de Kier Joffe, E.B.; Cadenas, E.; Boczkowski, J.; Carreras, M.C.; Poderoso, J.J. Tumor cell phenotype is sustained by selective MAPK oxidation in mitochondria. PLoS One 2008, 3, e2379. [Google Scholar] [CrossRef]
- Gergalova, G.; Lykhmus, O.; Komisarenko, S.; Skok, M. α7 nicotinic acetylcholine receptors control cytochrome c release from isolated mitochondria through kinase-mediated pathways. Int. J. Biochem. Cell Biol. 2014, 49, 26–31. [Google Scholar] [CrossRef]
- Chao, X.; Zhou, J.; Chen, T.; Liu, W.; Dong, W.; Qu, Y.; Jiang, X.; Ji, X.; Zhen, H.; Fei, Z. Neuroprotective effect of osthole against acute ischemic stroke on middle cerebral ischemia occlusion in rats. Brain Res. 2010, 1363, 206–211. [Google Scholar] [CrossRef]
- Chen, T.; Cao, L.; Dong, W.; Luo, P.; Liu, W.; Qu, Y.; Fei, Z. Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK pathway. Neurochem. Res. 2012, 37, 983–990. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, L.; Qu, Y.; Huo, K.; Jiang, X.; Fei, Z. The selective mGluR5 agonist CHPG protects against traumatic brain injury in vitro and in vivo via ERK and Akt pathway. Int. J. Mol. Med. 2012, 29, 630–636. [Google Scholar]
- Zhang, X.; Shi, M.; Bjoras, M.; Wang, W.; Zhang, G.; Han, J.; Liu, Z.; Zhang, Y.; Wang, B.; Chen, J.; et al. Ginsenoside Rd promotes glutamate clearance by up-regulating glial glutamate transporter GLT-1 via PI3K/AKT and ERK1/2 pathways. Front. Pharmacol. 2013, 4. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Wang, X.; Lau, W.; Xing, Y.; Zhang, X.; Ma, X.; Gao, F. Ginsenoside Rd attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3beta signaling and inhibition of the mitochondria-dependent apoptotic pathway. PLoS One 2013, 8, e70956. [Google Scholar]
- Sussman, M.A. Mitochondrial integrity: Preservation through Akt/Pim-1 kinase signaling in the cardiomyocyte. Expert Rev. Cardiovasc. Ther. 2009, 7, 929–938. [Google Scholar] [CrossRef]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediated mitochondrial protection in the heart: metabolic and survival pathways to the rescue. J. Bioenerg. Biomembr. 2009, 41, 169–180. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, M.; Ye, R.; Wang, W.; Liu, X.; Zhang, G.; Han, J.; Zhang, Y.; Wang, B.; Zhao, J.; et al. Ginsenoside Rd attenuates Tau protein phosphorylation via the PI3K/AKT/GSK-3β pathway after transient forebrain ischemia. Neurochem. Res. 2014. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, G.Y.; Ahlemeyer, B.; Pang, L.; Che, X.M.; Culmsee, C.; Klumpp, S.; Krieglstein, J. Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J. Neurosci. 2002, 22, 3898–3909. [Google Scholar]
- Kuroki, Y.; Fukushima, K.; Kanda, Y.; Mizuno, K.; Watanabe, Y. Neuroprotection by estrogen via extracellular signal-regulated kinase against quinolinic acid-induced cell death in the rat hippocampus. Eur. J. Neurosci. 2001, 13, 472–476. [Google Scholar] [CrossRef]
- Guerra, B.; Diaz, M.; Alonso, R.; Marin, R. Plasma membrane oestrogen receptor mediates neuroprotection against beta-amyloid toxicity through activation of Raf-1/MEK/ERK cascade in septal-derived cholinergic SN56 cells. J. Neurochem. 2004, 91, 99–109. [Google Scholar] [CrossRef]
- Cecatto, C.; Amaral, A.U.; Leipnitz, G.; Castilho, R.F.; Wajner, M. Ethylmalonic acid induces permeability transition in isolated brain Mitochondria. Neurotox. Res. 2014. [Google Scholar] [CrossRef]
- Yu, Z.; Liu, N.; Li, Y.; Xu, J.; Wang, X. Neuroglobin overexpression inhibits oxygen-glucose deprivation-induced mitochondrial permeability transition pore opening in primary cultured mouse cortical neurons. Neurobiol. Dis. 2013, 56, 95–103. [Google Scholar] [CrossRef]
- Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Chernyshov, V.; Kryukova, E.; Tsetlin, V.; Komisarenko, S.; Skok, M. Mitochondria express α7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: study on isolated mitochondria. PLoS One 2012, 7, e31361. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, J.-S.; Wang, J.-F.; He, B.-R.; Cui, Y.-S.; Fang, X.-Y.; Ni, J.-L.; Chen, J.; Wang, K.-Z. Ginsenoside Rd Attenuates Mitochondrial Permeability Transition and Cytochrome c Release in Isolated Spinal Cord Mitochondria: Involvement of Kinase-Mediated Pathways. Int. J. Mol. Sci. 2014, 15, 9859-9877. https://doi.org/10.3390/ijms15069859
Zhou J-S, Wang J-F, He B-R, Cui Y-S, Fang X-Y, Ni J-L, Chen J, Wang K-Z. Ginsenoside Rd Attenuates Mitochondrial Permeability Transition and Cytochrome c Release in Isolated Spinal Cord Mitochondria: Involvement of Kinase-Mediated Pathways. International Journal of Molecular Sciences. 2014; 15(6):9859-9877. https://doi.org/10.3390/ijms15069859
Chicago/Turabian StyleZhou, Jin-Song, Jiang-Feng Wang, Bao-Rong He, Yong-Sheng Cui, Xiang-Yi Fang, Jian-Long Ni, Jie Chen, and Kun-Zheng Wang. 2014. "Ginsenoside Rd Attenuates Mitochondrial Permeability Transition and Cytochrome c Release in Isolated Spinal Cord Mitochondria: Involvement of Kinase-Mediated Pathways" International Journal of Molecular Sciences 15, no. 6: 9859-9877. https://doi.org/10.3390/ijms15069859